Summary
Congenital immunodeficiency disorders are characterized by a deficiency, absence, or defect in one or more of the main components of the immune system. These disorders are genetically determined and typically manifest during infancy and childhood as frequent, chronic, or opportunistic infections. Classification is based on the component of the immune system that is deficient, absent, or defective. The diagnosis is confirmed with tests such as differential WBC count, absolute lymphocyte count, quantitative immunoglobulin (Ig) measurements, and antibody titers. Treatment usually consists of prophylactic antibiotics to manage and prevent infections. The prognosis in congenital immunodeficiency disorders is variable and depends on the specific disorder.
Overview
Congenital B-cell immunodeficiencies
| Overview of congenital B-cell immunodeficiencies | |||
|---|---|---|---|
| Immunodeficiency | Etiology | Clinical features | Diagnostic findings |
| X-linked (Bruton) agammaglobulinemia |
|
|
|
| Selective IgA deficiency |
|
|
|
| Common variable immunodeficiency |
|
|
|
Congenital T-cell immunodeficiencies
| Overview of congenital T-cell immunodeficiencies | |||
|---|---|---|---|
| Immunodeficiency | Etiology | Clinical features | Diagnostic findings |
| DiGeorge syndrome |
|
|
|
| Autosomal dominant hyper-IgE syndrome (Job syndrome) |
|
|
|
| IL-12 receptor deficiency |
|
|
|
| Chronic mucocutaneous candidiasis |
|
|
|
| IPEX syndrome |
|
|
|
Congenital combined immunodeficiencies
| Overview of congenital combined immunodeficiencies | |||
|---|---|---|---|
| Immunodeficiency | Etiology | Clinical features | Diagnostic findings |
| Severe combined immunodeficiency |
|
|
|
| Wiskott-Aldrich syndrome (WAS) |
|
|
|
| Hyper-IgM syndrome |
|
|
|
| Ataxia telangiectasia |
|
|
|
Congenital neutrophil and phagocyte disorders
| Overview of congenital neutrophil and phagocytes disorders | |||
|---|---|---|---|
| Immunodeficiency | Etiology | Clinical features | Diagnostic findings |
| Chronic granulomatous disease (CGD) |
|
|
|
| Leukocyte adhesion deficiency type 1 |
|
|
|
| Chédiak-Higashi syndrome |
|
|
|
| Myeloperoxidase deficiency |
|
|
|
| Severe congenital neutropenia |
|
|
|
Complement disorders
| Overview of complement disorders | ||||
|---|---|---|---|---|
| Immunodeficiency | Etiology | Clinical features | Diagnostic findings | |
| C1 esterase inhibitor deficiency |
|
|
|
|
| Early complement deficiencies | C1, C2, and C4 deficiency |
|
|
|
| C3 deficiency |
|
|
||
| Terminal complement deficiency |
|
|
||
Congenital B-cell immunodeficiencies
B-cell defects (humoral immunity deficiencies) account for 50–60% of all primary immunodeficiencies.
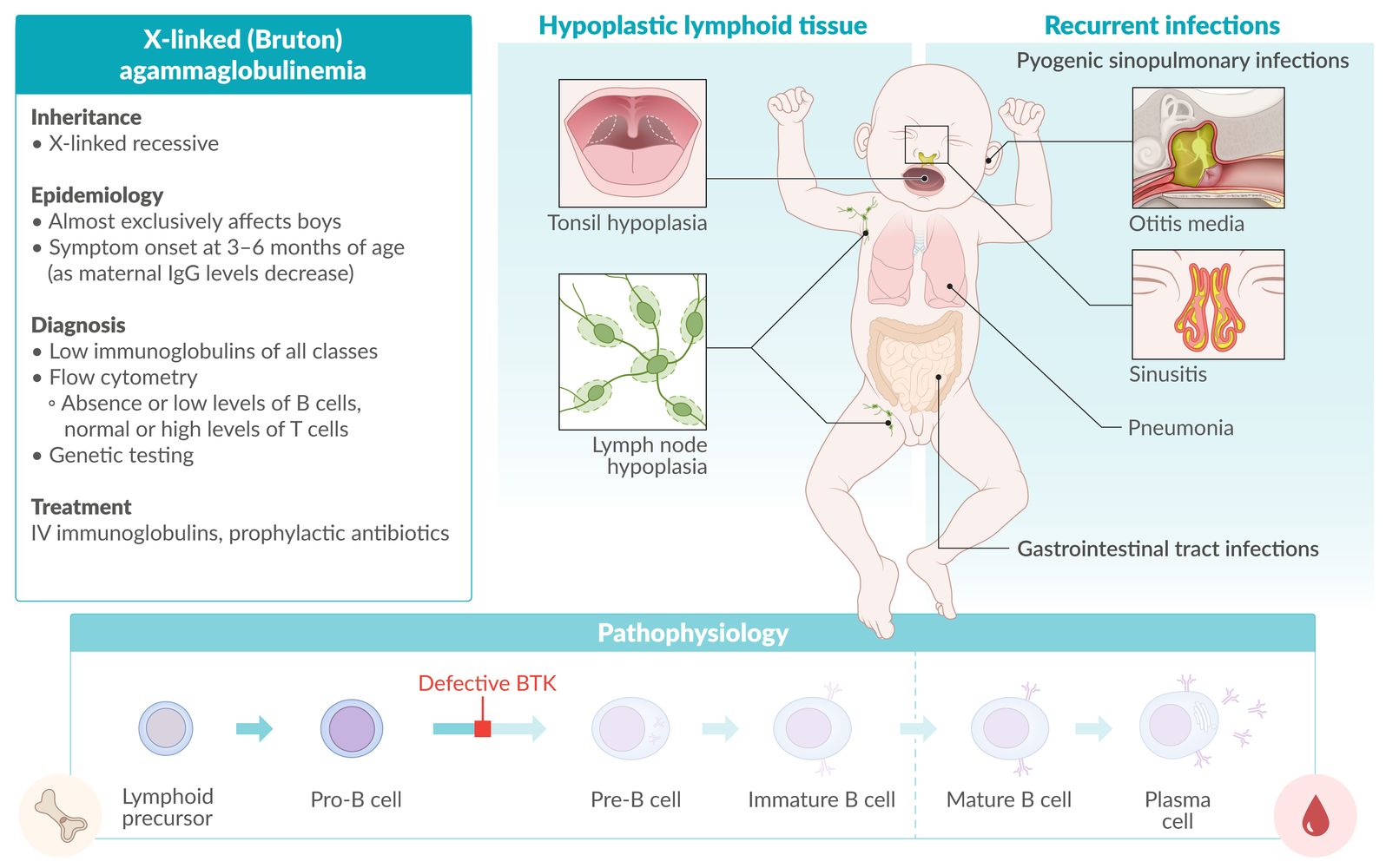
X-linked (Bruton) agammaglobulinemia [3]
- Definition: X-linked recessive; disease that causes a complete deficiency of mature B lymphocytes
- Epidemiology: occurs mainly in boys
- Etiology: defect of Bruton tyrosine kinase (BTK) expressed in B cells → complete deficiency of mature B cells
-
Clinical features: Symptoms develop between 3 and 6 months of age when maternal IgG levels in fetal serum start to decrease.
- Hypoplasia of lymphoid tissue (e.g., tonsils, lymph nodes)
- Recurrent, severe, pyogenic infections (e.g., pneumonia, otitis media), especially with encapsulated bacteria (S. pneumoniae, N. meningitidis, and H. influenzae)
- Hepatitis virus and enterovirus (e.g., Coxsackie virus) infections
-
Diagnosis
-
Flow cytometry
- Absent or low levels of B cells (marked by CD19, CD20, and CD21)
- Normal or high T cells
- Low immunoglobulins of all classes
- Absent lymphoid tissue, i.e., no germinal centers and primary follicles
-
Flow cytometry
-
Treatment
- IV immunoglobulins
- Prophylactic antibiotics

Live vaccines (e.g., MMR) are contraindicated in patients with X-linked (Bruton) agammaglobulinemia.
X-linked (Bruton) agammaglobulinemia: Brutal defects in a B cell make little Boys feel unwell.
Selective IgA deficiency (SIgAD) [4][5]
- Definition: most common primary immunodeficiency; that is characterized by a near or total absence of serum and secretory IgA
- Epidemiology: approx. 1:220 to 1:1,000
- Etiology: unknown
-
Clinical features
- Often asymptomatic
- May manifest with sinusitis or respiratory infections (S. pneumoniae, H. influenzae)
- Chronic diarrhea, partially due to elevated susceptibility to parasitic infection; (e.g. by Giardia lamblia)
- Associated with autoimmune diseases (e.g., gluten-sensitive enteropathy, inflammatory bowel disease, immune thrombocytopenia) and atopy
- Anaphylactic reaction to products containing IgA (e.g., intravenous immunoglobulin)
-
Diagnosis
- Decreased serum IgA levels (< 7 mg/dL)
- Normal IgG and IgM levels
- False-positive pregnancy tests [6]
-
Treatment
- Treatment of infections
- Prophylactic antibiotics
- Intravenous infusion of IgA is not recommended because of the risk of anaphylactic reactions (caused by the production of anti-IgA antibodies).
To prevent transfusion reactions, IgA-deficient patients must be given washed blood products without IgA or obtain blood from an IgA-deficient donor.
The Six A's of selective IgA deficiency: Asymptomatic, Airway infections, Anaphylaxis to IgA-containing products, Autoimmune diseases, Atopy
Common variable immunodeficiency (CVID) [7][8][9]
- Definition: primary immunodeficiency with low serum levels of all immunoglobulins despite phenotypically normal B cells
-
Epidemiology
- Sex: ♀ = ♂
- Onset: later than other B-cell defects (typically at 20–40 years of age) [10]
- Etiology: Most cases are sporadic with no known family history.
- Pathophysiology: B cells are phenotypically normal but are unable to differentiate into Ig-producing cells, resulting in low immunoglobulins of all classes.
-
Clinical features
- Recurrent pyogenic respiratory infections, e.g., sinopulmonary infections (in rare cases, enteroviral meningitis)
- Associated with a high risk of lymphoma, gastric cancer, bronchiectasis, and autoimmune disorders (e.g., rheumatoid arthritis, autoimmune hemolytic anemia, immune thrombocytopenia, vitiligo) [11]
-
Diagnosis
- Quantitative immunoglobulin levels: low levels of IgG, IgA, and IgM
- Decreased number of plasma cells
- Flow cytometry shows subsets of normal B and T cells
- Poor response to immunizations
-
Treatment
- Treatment of infections
- Prophylactic antibiotics
- IV immunoglobulins
Transient hypogammaglobulinemia of infancy (THI) [12][13]
- Definition: an age-related delay in immunoglobulin production despite having normal levels of B cells
-
Epidemiology
- Approx. 1:1000 live births [13]
- Onset: > 6 months of age; Ig levels typically increase and normalize at 2–6 years of age. [12]
-
Clinical features
- Recurrent infections (most common: sinopulmonary infections, otitis media, bronchitis, tonsillitis, diarrhea)
- Atopic manifestations (e.g., asthma, food allergies)
- Failure to thrive and/or developmental delay
- Can be asymptomatic
- Severe and/or invasive infections, such as meningitis and bacteremia (rare)
-
Diagnosis
-
Quantitative immunoglobulin levels
- IgG is ≤ 2 SD of age-appropriate levels.
- Levels of IgA and IgM may be low.
- Flow cytometry shows subsets of normal B and T cells.
-
Presence of specific antibodies
- Post-exposure IgG antibodies (e.g., rubella, varicella)
- Post-immunization IgG antibodies (e.g., tetanus toxoid, pneumococcal polysaccharide)
- Isohemagglutinins (anti-A, anti-B)
-
Quantitative immunoglobulin levels
-
Treatment
- Prophylactic antibiotics in patients with recurrent infections
- IV immunoglobulins in patients who develop severe and/or invasive infections or recurrent infections despite prophylaxis
- Appropriate treatment in patients with atopic manifestations (e.g., bronchodilators)
- Routine immunization schedule should be continued.
BTK: B-cell tyrosine kinase
© AMBOSS
Congenital T-cell immunodeficiencies
T-cell defects (cellular immunity deficiencies) are responsible for 5–10% of congenital immunodeficiencies.
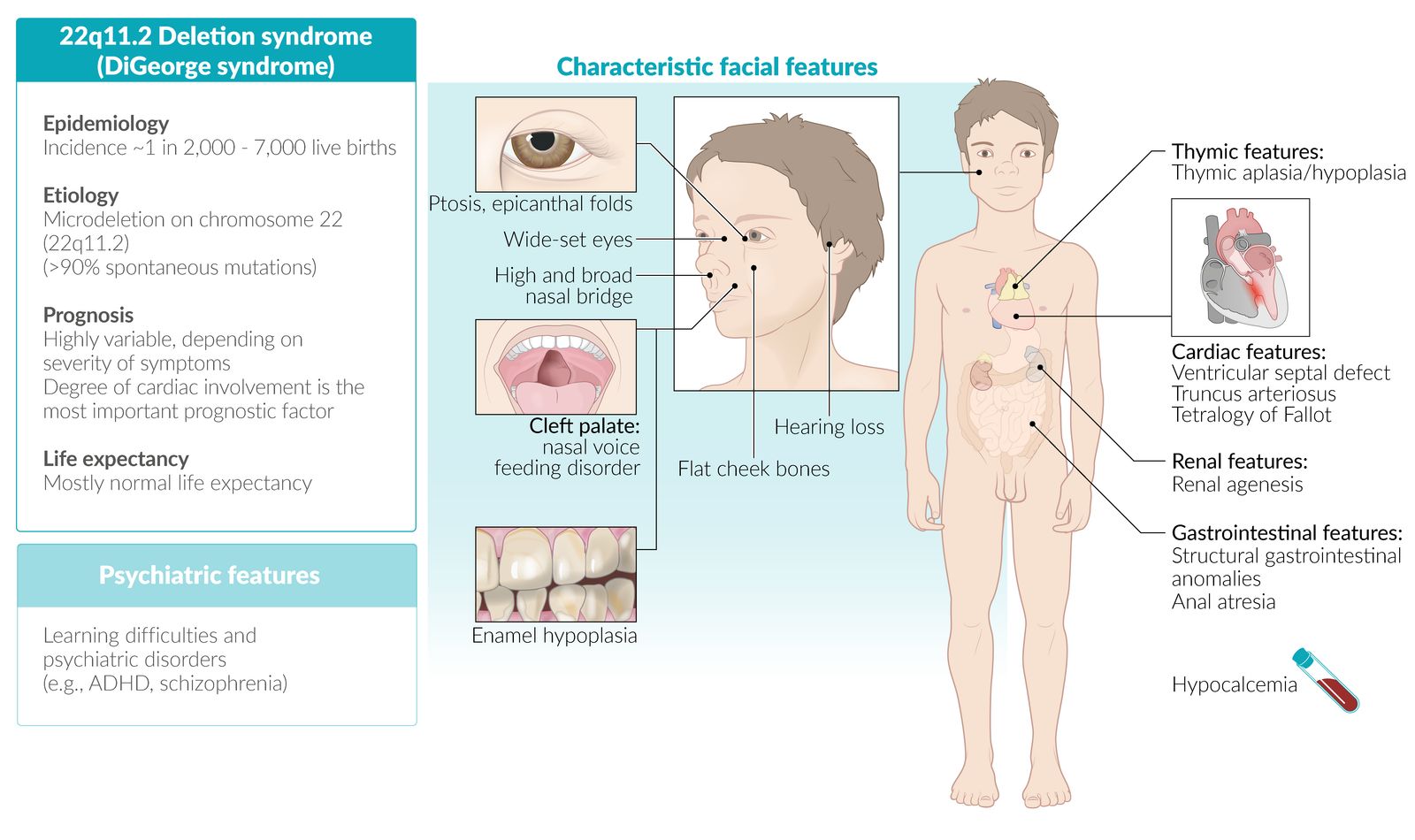
DiGeorge syndrome (22q11.2 deletion syndrome) [14]
- Definition: genetic disorder, which is characterized by a microdeletion at the 22q11.2 region resulting in a range of developmental abnormalities
- Epidemiology: ∼ 1 in 2,000 to 7,000 live births
- Etiology: microdeletion at chromosome 22 (22q11.2); de novo mutation (∼ 90%), autosomal dominant (∼ 10%)
-
Pathophysiology
- The TBX1 gene in the 22q11.2 region is the main gene responsible for the DiGeorge phenotype
- Deletions in TBX1 lead to malformations in the pharyngeal apparatus, which results in abnormalities of the:
- Bones and muscles of the face and neck → abnormal facies, cleft palate
- Outflow tract of the heart → heart defects (e.g., ToF)
- 3rd and 4th pharyngeal pouches → thymus hypoplasia (T-cell immunodeficiency) and parathyroid hypoplasia (hypocalcemia)
-
Clinical features
-
Cardiac anomalies in DiGeorge syndrome
- Conotruncal abnormalities (e.g., tetralogy of Fallot or persistent truncus arteriosus)
- Ventricular septal defect (VSD)
- Atrial septal defect (ASD)
-
Anomalous face
- Prominent nasal bridge
- Hypoplastic wing of the nose
- Dysplastic ears
- Micrognathia; (small lower jaw) and/or retrognathia
- Thymus aplasia/hypoplasia: recurrent infections (viral/fungal/PCP pneumonia) due to T-cell deficiency
- Cleft palate
- Hypoparathyroidism: hypocalcemia with tetany
-
Cardiac anomalies in DiGeorge syndrome
-
Diagnosis
- Detection of 22q11.2 deletion via fluorescence in situ hybridization (FISH)
- ↓ PTH and Ca2+
- ↓ Absolute T-lymphocyte count
- Delayed hypersensitivity skin testing
- CXR: absence of thymic shadow
-
Treatment
-
Immune deficiency treatment
- Antibiotics, virostatics, and antimycotics
- PCP prophylaxis
- Consider bone marrow transplant and/or IVIG
- Possible thymus transplantation
-
Immune deficiency treatment
- Prognosis: mostly normal life expectancy with appropriate medical care
CATCH-22 is the acronym for typical features of DiGeorge syndrome: Cardiac anomalies; Anomalous face; Thymic aplasia/hypoplasia; Cleft palate; Hypocalcemia; Chromosome 22.
 fact sheet")


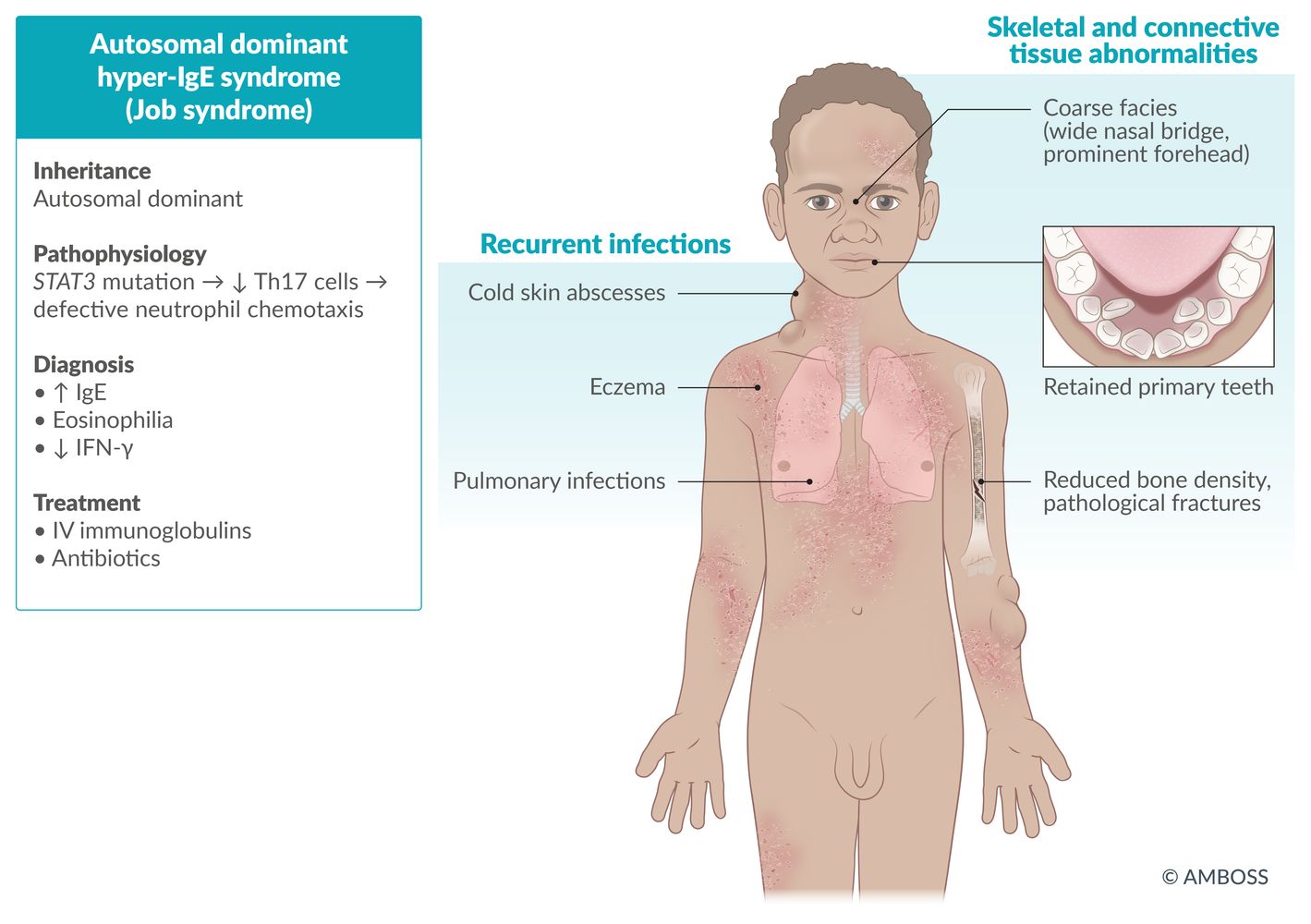
Autosomal dominant hyperimmunoglobulin E syndrome (Job syndrome) [15]
- Definition: defect in neutrophil chemotaxis
- Etiology: autosomal dominant; STAT3 mutation → ↓ Th17 cells → ↓ neutrophil chemotaxis
-
Clinical features
- Coarse Facies
- Recurrent, cold Abscesses, recurrent bacterial (staphylococcal) infections
- Retained primary Teeth
- Hyper-IgE (Eosinophilia)
- Dermatologic (severe eczema)
- Other features: Decreased bone density increases the risk of bone fractures following minor trauma. [16]
-
Diagnosis
- ↑ IgE
- (Variable) eosinophilia
- ↓ IFN γ
-
Treatment
- Antibiotics and prophylaxis (with penicillinase-resistant antibiotics)
- IV immunoglobulin therapy
FATED is the acronym for the typical features of Autosomal dominant hyper-IgE syndrome: Coarse Facies/Fractures; Abscesses; Retained primary Teeth; Hyper-IgE/Eosinophilia); Dermatologic (severe eczema).

IL-12 receptor deficiency [17][18][19]
- Definition: impaired Th1 response due to ↓ IL-12 receptors
- Etiology: autosomal recessive
-
Pathophysiology
- Normally, antigen-presenting macrophages release IL-12 → Th cells transformation to T1 type → release of IFN-γ to activate macrophages
- Defective IL-12 receptors: macrophages cannot be activated by IFN-γ → no cytotoxicity in cells infected with intracellular pathogens (e.g., Mycobacteria, Salmonella)
-
IL-12 receptor deficiency is the underlying pathology in most cases of high Mendelian susceptibility to mycobacterial disease (MSMD)
- Affected individuals have defects in IFN-γ mediated immunity.
- This increases susceptibility to infection with weakly virulent mycobacteria (e.g., environmental mycobacteria, BCG vaccine).
-
Clinical features
- The age of onset varies (depends on the age at exposure to causative pathogens): ∼ 1–3 years of age
-
Features of disseminated disease
- Mycobacterial infections (e.g., developing tuberculosis after BCG vaccination)
- Fungal infections
- Diagnosis: : ↓ IFN-γ
- Treatment: antibiotics and IFN-γ therapy
Chronic mucocutaneous candidiasis
- Definition: impaired or absent T-cell response to Candida antigens
-
Etiology
- Several congenital defects that result in impaired T-cell function
- Autoimmune regulator (AIRE protein) deficiency is a common cause.
- The majority of cases are caused by a gain of function mutation in the STAT1 gene. [20]
- Pathophysiology: defects in IL-17 and IL-17 receptors → insufficient cell-mediated immune response to Candida infections
-
Clinical features
- Persistent or, less commonly, recurrent noninvasive Candida infections of the skin, mucous membranes, and nails
- Associated autoimmune disorders (e.g., immune thrombocytopenic purpura, rheumatoid arthritis)
-
Diagnosis
- Usually clinical
- Absent in vitro T-cell proliferation when exposed to Candida antigen
- Absent cutaneous reaction to Candida antigen
-
Treatment
- Antifungal therapy
- Treatment of associated conditions
IPEX syndrome (Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked)
- Definition: : A syndrome characterized by a dysfunctional regulatory T-cell lineage that leads to autoimmunity as a result of disrupted tolerance to self-antigens.
-
Etiology
- X-linked recessive type of inheritance
- Mutation in transcription factor FOXP3 → unchecked activation of T cells against host tissues
-
Clinical features
- Classically presents in early infancy
- Lymphadenopathy and/or chronic lymphoid tissue hypertrophy (e.g., enlarged tonsils)
- Eczema, possibly accompanied by other autoimmune dermatologic conditions
- Autoimmune endocrine conditions (e.g., hyperthyroidism or hypothyroidism, type 1 diabetes mellitus in male individuals)
- Enteropathy, manifesting as e.g., chronic diarrhea
- Nail dystrophy
- Failure to thrive
-
Diagnosis
- Primarily based on clinical examination and family history
- Genetic testing: mutations in FOXP3
- Flow cytometry: severely reduced or absent CD4+CD25+ regulatory T cells with otherwise normal T-cell populations
-
Treatment
- Nutritional support
- Immunosuppression: tacrolimus, cyclosporine, or sirolimus
- Rituximab
- Bone marrow transplant
© AMBOSS
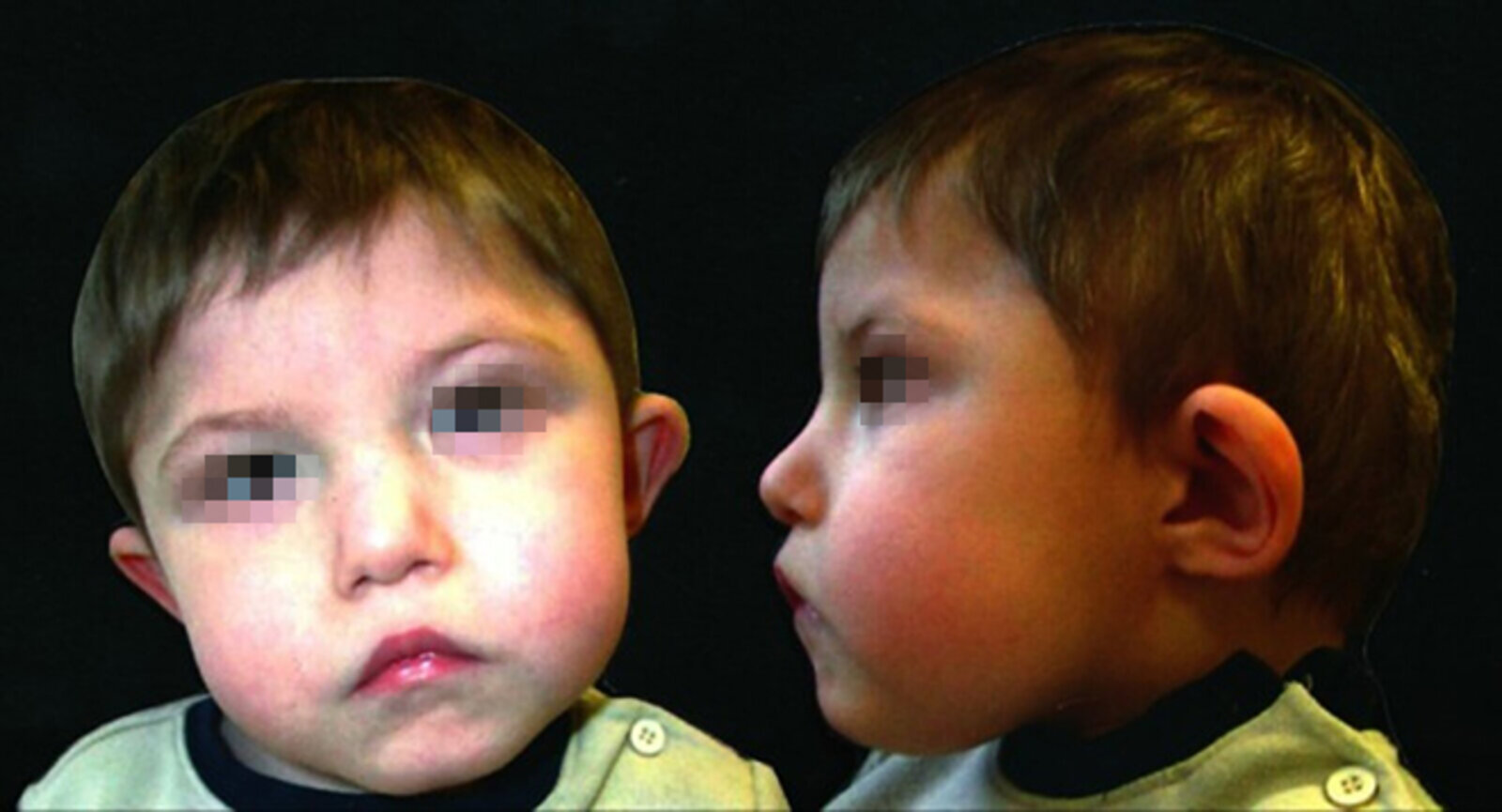
Broad nasal bridge and nasal tip, ocular hypertelorism, dysplastic and low-set ears as well as micrognathia.
Source: “Figure 1, in: Towards a safety net for management of 22q11.2 deletion syndrome: guidelines for our times” by A. Habel, R. Herriot, D. Kumararatne et al., Springer Link, licensed under CC BY 4.0. Modifications: person anonymized.

Original title: “Genetics: Section 12.3 - DiGeorge Syndrome”. Created by: Physeo™.
© AMBOSS
Congenital mixed immunodeficiencies
Severe combined immunodeficiency (SCID, Glanzmann–Riniker syndrome, alymphocytosis)
- Definition: : A rare genetic condition caused by numerous genetic mutations that result in the defective development of functional B cells and T cells.
-
Etiology: various mutations, the most common of which are:
- X-linked recessive: mutations in the gene encoding the common gamma chain → defective IL-2R gamma chain receptor linked to JAK3 (most common SCID mutation)
-
Autosomal recessive
- Adenosine deaminase (ADA) deficiency → accumulation of toxic metabolites (deoxyadenosine and dATP) and disrupted purine metabolism → accumulation of dATP inhibits the function of ribonucleotide reductase → impaired generation of deoxynucleotides
- Janus-associated kinase 3 (JAK3) deficiency
- RAG mutation results in faulty VDJ recombination (see “Immunoglobulin properties”).
-
Clinical features
- Normal at birth
- Severe, recurrent infections: bacterial diarrhea, chronic candidiasis (thrush), viral and protozoal infections
- Failure to thrive
- Chronic diarrhea
- Lymph nodes and tonsils may be absent
-
Subtype: Omenn syndrome (an autosomal recessive form of SCID) [21]
- Etiology: mutations of RAG gene or ILRA7 gene
- Clinical features
- Onset before 3 months of age
- Erythroderma, adenopathy, and hepatosplenomegaly
- Severe, recurrent infections
- Failure to thrive
- Diagnosis: oligoclonal T cells, eosinophilia, ↑ IgE levels
-
Diagnosis
- Quantitative PCR: ↓ T-cell receptor excision circles (TRECs); detection of TRECs is used in the newborn screening for SCID
- Flow cytometry: absent T cells
- CXR: absent thymic shadow
- Lymph node biopsy: absent germinal centers
-
Treatment
- IV immunoglobulins
- PCP prophylaxis
- Bone marrow transplant or stem cell transplantation
- Avoidance of live vaccines
- Prognosis: often fatal in the first year of life if left untreated [22]
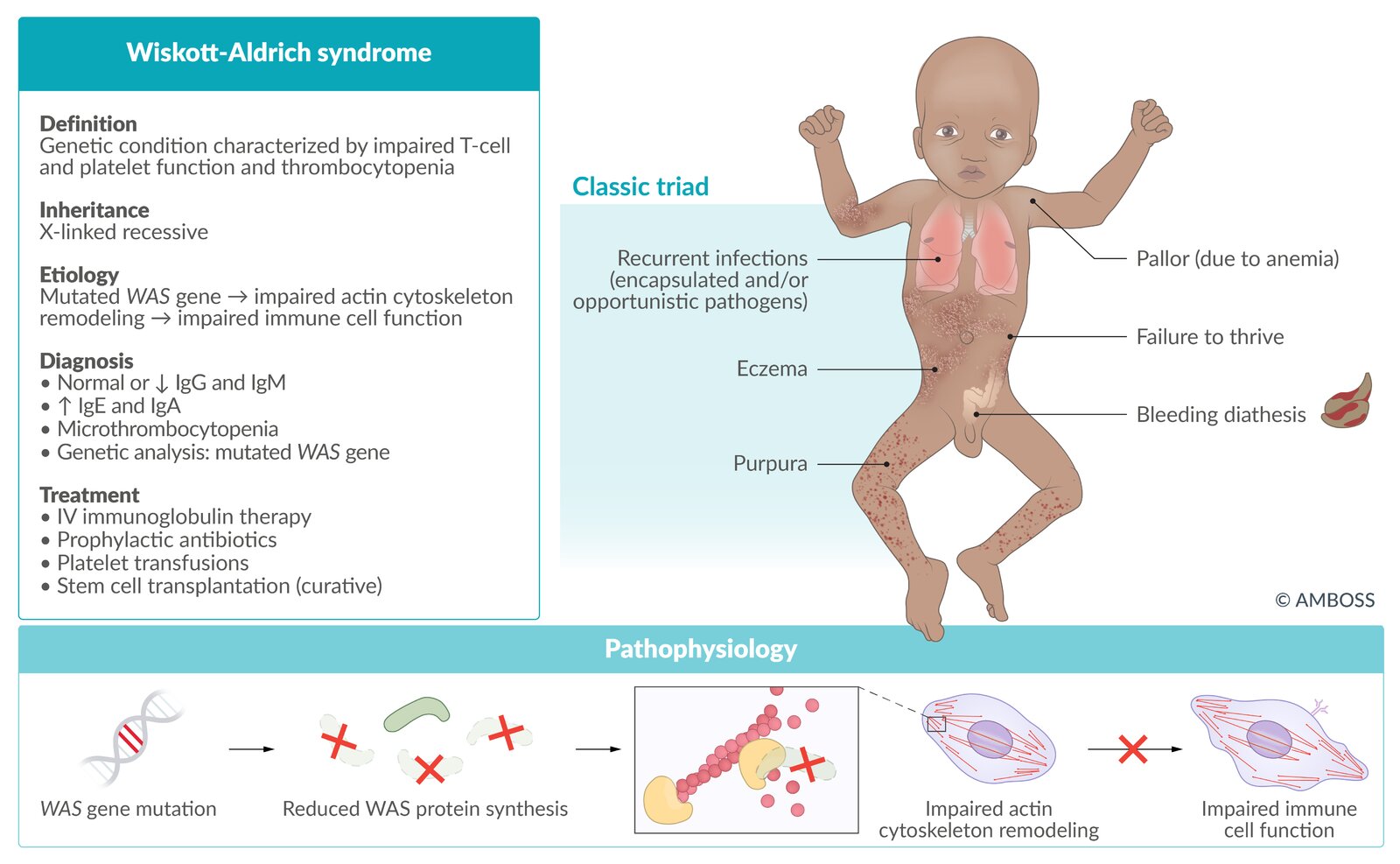
Wiskott-Aldrich syndrome (WAS)
- Definition: genetic condition characterized by impaired function of T cells and thrombocytopenia
- Epidemiology: : occurs primarily in males
- Etiology: mutated WAS gene (X-linked recessive inheritance) → impaired signaling to actin cytoskeleton reorganization → defective antigen presentation
-
Clinical features
- Onset of symptoms: from birth
- Classic triad
- Purpura (bleeding diathesis)
- Eczema (high risk of atopic disorders)
- Recurrent opportunistic infections with encapsulated organisms; in the first years of life (e.g., otitis media)
- Increased risk of autoimmune diseases and hematological malignancies (e.g., lymphoma, leukemia)
-
Diagnosis
- Normal or ↓ IgG and IgM
- ↑ IgE and IgA
- Thrombocytopenia with small platelets
- Genetic analysis (confirmatory test): mutated WAS gene
-
Treatment
- IV immunoglobulin therapy
- Prophylactic antibiotics
- Platelet transfusions
- Stem cell transplantation may be curative.
- Prognosis: : shortened life expectancy


Classic findings of Wiskott-Aldrich syndrome (Wiskott-Aldrich syndrome, Purpura, Eczema, Recurrent infections): WisPER
Hyper-IgM syndrome [23]
- Definition: A group of syndromes characterized by impaired interaction between Th cells and B cells that results in a B cell class-switching defect
- Epidemiology: CD40 ligand deficiency is the most common form.
-
Etiology
- X-linked recessive inheritance
- CD40L deficiency in Th cells leads to abnormal interaction with B cells that express the CD40 receptor, disrupting B-cell class-switching, which leads to depleted immunoglobulins
-
Clinical features
- Recurrent severe pyogenic infections that manifest since childhood
- Opportunistic sinopulmonary infections (commonly Pneumocystis jirovecii and Histoplasma)
- Cryptosporidium enteritis (which may lead to biliary disease, cirrhosis, and cholangiocarcinoma)
- CMV hepatitis
- Failure to thrive
- Recurrent severe pyogenic infections that manifest since childhood
-
Diagnosis
- ↓ IgG, IgA, IgE
- Normal or ↑ IgM
- Lymph node biopsy: absent germinal centers
-
Treatment
- IV immunoglobulin therapy
- Prophylactic antibiotics
- Recombinant human granulocyte-colony stimulating factor
- Stem cell transplantation may be curative.
Ataxia telangiectasia
- See “Ataxia telangiectasia” for details.
© AMBOSS
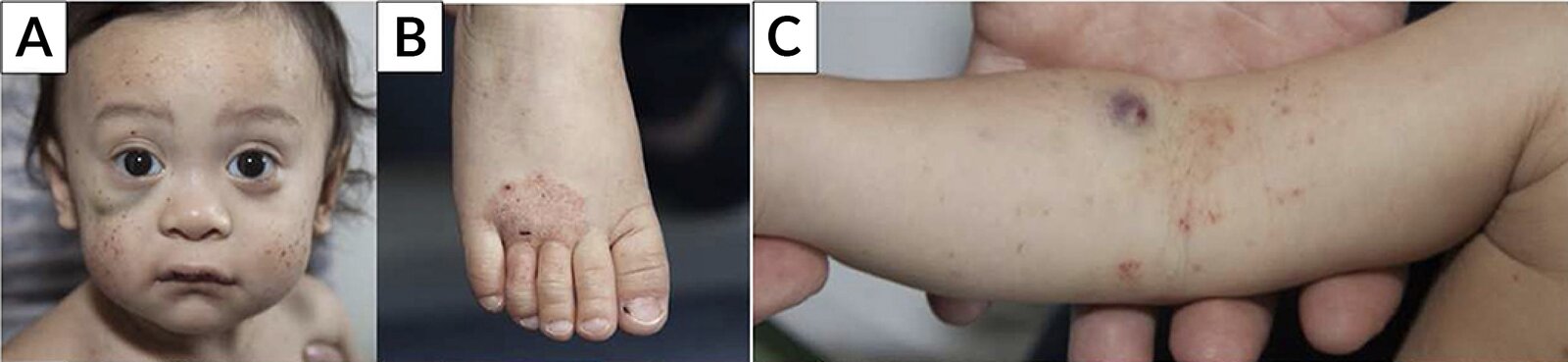
Face (A), foot (B), and right arm (C) of an 18-month-old boy
Multiple petechiae and hematomas are visible. Eczema is seen on the dorsal aspect of the right distal foot and the right antecubital fossa.
The combination of purpura and eczema in a young boy suggests Wiskott-Aldrich syndrome (also known as "eczema-thrombocytopenia-immunodeficiency syndrome"). The condition is X-linked recessive and therefore occurs primarily in male individuals. Affected individuals are also at increased risk for recurrent pyogenic infections.
Source: “Fig 1A/C/E, in: Wiskott-Aldrich Syndrome (WAS) and Dedicator of Cytokinesis 8- (DOCK8) Deficiency” by Albert MH, Freeman AF, Frontiers in Pediatrics, licensed under CC BY 4.0. Modifications: image cropped; removal of the letters A/C/E & their replacement with Lato letters A/B/C.
Congenital neutrophil and phagocyte disorders
Phagocytic defects are characterized by the impaired ability of phagocytic cells (e.g., monocytes, macrophages, granulocytes such as neutrophils and eosinophils) to kill pathogens. These types of defects account for 10–15% of primary immunodeficiencies.
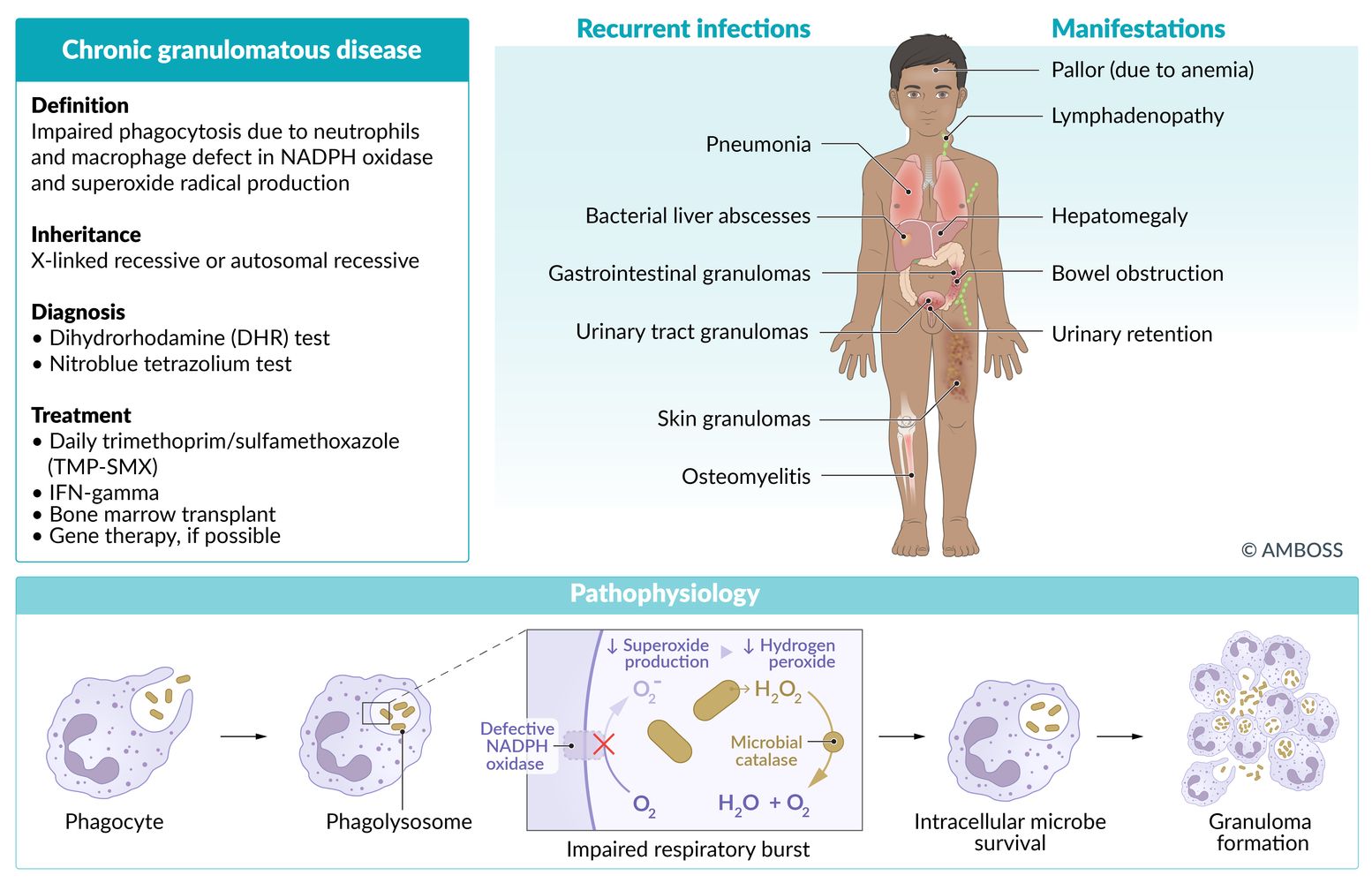
Chronic granulomatous disease (CGD)
- Definition: deficiency of superoxide production by polymorphonuclear neutrophils and macrophages
-
Etiology
- X-linked recessive or autosomal recessive inheritance (2:1)
-
Defective phagocytic nicotinamide adenine dinucleotide phosphate (NADPH) oxidase that results in:
- Defective reactive oxygen species (ROS) production (e.g., superoxide) → impaired ability to deactivate or kill ingested microorganisms
- Decreased respiratory burst in neutrophils
-
Clinical features
- Recurrent, severe infections (chronic skin, lymph node, bone, respiratory, GI, and urinary tract infections) with catalase-positive organisms (S. aureus, Nocardia spp., Escherichia coli, Candida, Klebsiella, Pseudomonas, Aspergillus, Serratia)
- Lymphadenopathy
- Granulomas of the skin and GI/GU tract
-
Diagnosis
-
Neutrophil assay
- Dihydrorhodamine test (DHR): flow cytometry test showing abnormal NADPH oxidase activity; (inability to metabolize dihydrorhodamine to fluorescent product, rhodamine → decreased green fluorescence) [24]
- Nitroblue tetrazolium dye reduction test: negative, i.e., incubated leukocytes fail to turn blue when exposed to nitroblue tetrazolium
- Hypergammaglobulinemia
- Anemia
- Genotyping is confirmatory
-
Neutrophil assay
-
Treatment
- Treatment of infections
- Life-long prophylactic antibiotics, e.g., TMP-SMX (for catalase-positive infections)
- Antifungal prophylaxis (e.g., itraconazole, voriconazole, posaconazole)
- Glucocorticoids for severe inflammation
- IFN-γ therapy
- Bone marrow transplant
- Possibly gene therapy

Leukocyte adhesion deficiency type 1 (LAD1)
- Definition: : A genetic condition characterized by a defect in the leukocytic chemotaxis that results in decreased phagocyte activity
-
Etiology
- Autosomal recessive inheritance
- Absence of the β2-integrin leukocyte adhesion surface molecule LFA-1 (CD18) prevents leukocytes from migrating to tissues during infection or inflammation.
-
Clinical features
- Recurrent nonsuppurative bacterial infections (e.g., skin and mucosal infections) with minimal inflammation due to dysfunctional neutrophils
- Impaired wound healing
- Omphalitis
- Delayed separation of the umbilical cord (> 30 days postpartum)
-
Diagnosis
- Leukocytosis; however, neutrophils are absent at the site of infection
- Flow cytometry: absent CD18, CD11a, CD11b, and CD11c
-
Treatment
- Prevention of further infections (e.g., adequate dental hygiene)
- Treatment of infections
- Bone marrow transplant
Chédiak-Higashi syndrome
- Definition: defect in neutrophil chemotaxis and microtubule polymerization dysfunction → defective phagosome-lysosome fusion
- Etiology: autosomal recessive; defective lysosomal trafficking regulator (LYST) gene
-
Clinical features
-
Recurrent pyogenic infections
- May be extensive if massive infiltration occurs
- Pathogens include: S. pyogenes, S. aureus, and Pseudomonas spp.
- Partial albinism
- Progressive degeneration of neurons and peripheral neuropathy
- Hemophagocytic lymphohistiocytosis (can occur in the accelerated phase and is potentially fatal)
-
Recurrent pyogenic infections
-
Diagnosis
- Peripheral smear shows giant cytoplasmic granules in granulocytes and platelets
- Mild coagulation abnormalities
- Pancytopenia (especially neutropenia)
- Treatment: bone marrow transplant

Classic findings of Chediak-Higashi syndrome (Albinism, Lymphohistiocytosis, Peripheral neuropathy, Infections, Neurodegeneration, Neutropenia): ALPINe
Myeloperoxidase deficiency
- Definition: : A genetic condition characterized by the deficiency or absence of myeloperoxidase enzyme in phagocytes that are unable to form hypochlorous acid (HClO) but have preserved respiratory burst (since NADPH oxidase is intact).
- Etiology: autosomal recessive mutation in the MPO gene
-
Clinical features
- Recurrent Candida infections (e.g., oral thrush, vulvovaginitis)
- Many patients are asymptomatic.
-
Diagnosis
- Positive nitroblue tetrazolium test: intact NADPH oxidase
- Absent myeloperoxidase on staining
- Mutations in MPO gene on sequencing
-
Treatment
- No specific treatment or prophylaxis
- Treatment of fungal infections
Severe congenital neutropenia
- Definition: A deficiency of neutrophils that occurs at or around birth due to bone marrow failure of the myeloid lineage.
-
Etiology
- Autosomal dominant, autosomal recessive, or X-linked recessive inheritance
- Mutations in the neutrophil elastase gene, HAX1 gene, and Wiskott-Aldrich syndrome gene
-
Clinical features
- Recurrent bacterial infections, e.g., gingivitis, otitis media, respiratory infections, and cellulitis due to Streptococcus and Staphylococcus
- Oral ulcerations are commonly seen by the age of 2 years.
-
Diagnosis
- Absolute neutropenia with relative monocytosis. Absolute neutrophil count is usually < 200/μL in infants.
-
Bone marrow examination
- Normal or decreased cellularity
- Arrest of the myeloid lineage at the promyelocyte/myelocyte stage
-
Treatment
- G-CSF
- Hematopoietic cell transplantation
© AMBOSS
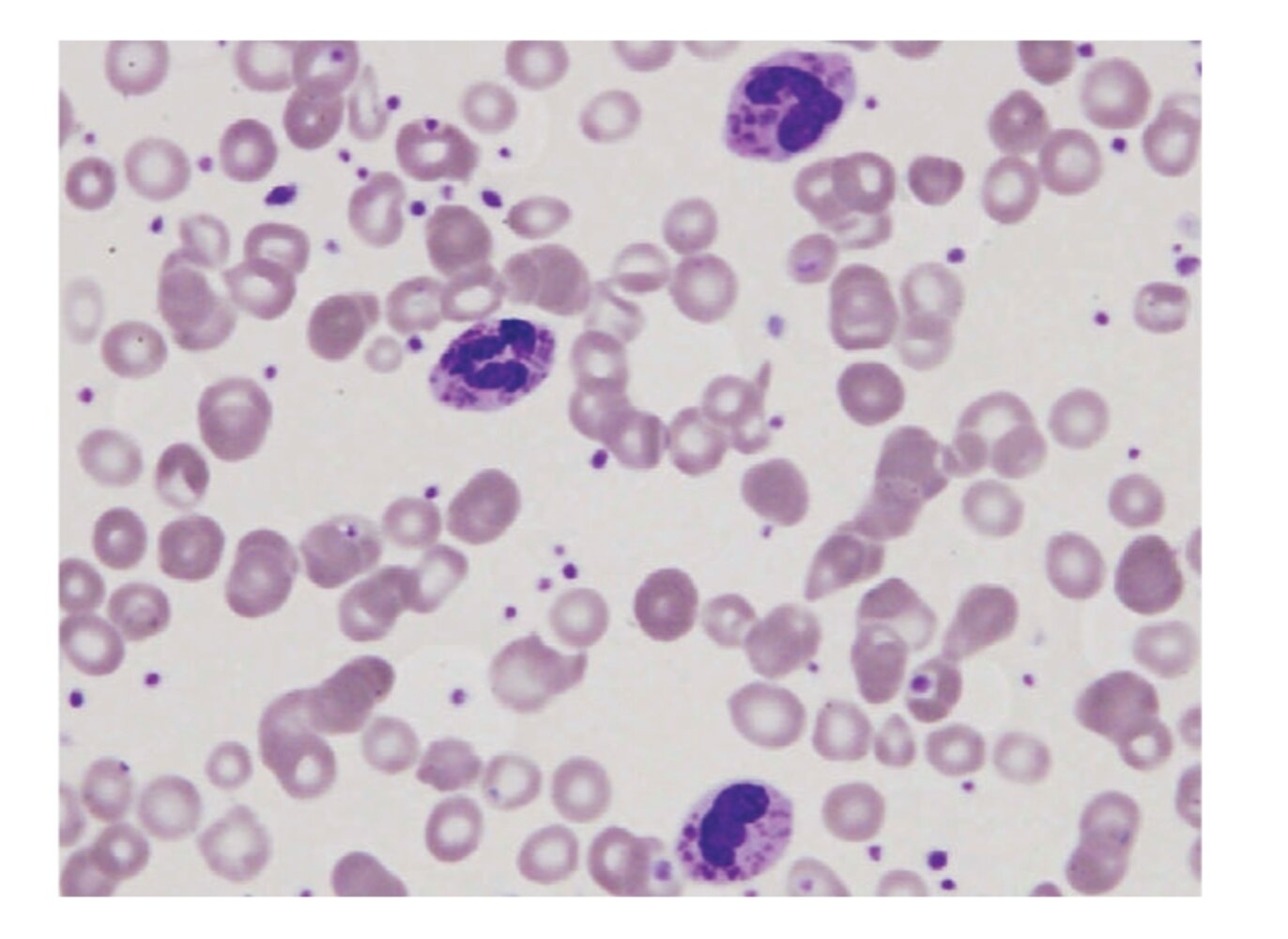
Photomicrograph of peripheral blood smear (Wright-Giemsa stain; very high magnification)
Inclusion bodies (examples indicated by arrows) and polymorphous nuclei (green overlay) can be seen within the leukocytes.
These findings are consistent with Chediak-Higashi syndrome.
Source: “Figure 2, in: Two Novel Mutations Identified in an African-American Child with Chediak-Higashi Syndrome” by Kerry Morrone, Yanhua Wang, Marjan Huizing, Elie Sutton, James G. White, William A. Gahl, and Karen Moody, Case Reports in Medicine, licensed under CC BY 3.0. The supplementary image with overlays of relevant areas was adapted from the image mentioned above (© AMBOSS).
Congenital complement deficiencies
Complement component deficiencies
Complement component deficiencies are a group of rare inherited genetic conditions characterized by absent or abnormal complement component proteins, which increases the risk of recurrent bacterial infections and autoimmune conditions.
| Overview of complement component deficiencies | ||||||
|---|---|---|---|---|---|---|
| Classification | Classical pathway component deficiencies | Alternative pathway component deficiencies | Terminal complement deficiency (C5–C9) [25] | |||
| C1q deficiency [26][27] | C1r deficiency and/or C1s deficiency | C2 deficiency [28] | C3 deficiency [29] | C4 deficiency [30] | ||
| Epidemiology |
|
|
|
|
|
|
| Etiology |
|
|
|
|||
|
|
|
|
|||
| Pathophysiology |
|
|
|
|
|
|
| Clinical features |
|
|||||
|
|
|
|
|
|
|
| Diagnosis |
|
|||||
| Management |
|
|||||
| Complications [2] |
|
|
||||
|
|
|
|
|
||
C1 esterase inhibitor deficiency (hereditary angioedema)
- See “Bradykinin-mediated angioedema.”
Mannose-binding lectin deficiency (MBL deficiency) [35]
- Definition: an inherited genetic condition characterized by low levels of MBL associated with increased susceptibility to infections
- Epidemiology: common; affects an estimated 10–30% of the population worldwide
- Etiology: mutations in the MBL2 gene
- Pathophysiology: genetic mutations → ↓ production of MBL subunits or impaired subunit assembly into functional MBL → ↓ activation of the lectin complement pathway → ↑ susceptibility to infections [36]
- Clinical features: symptoms of recurrent infections
-
Diagnostics
- Measurement of serum MBL protein levels
- Confirmatory molecular genetic testing
- Management and complications: see C1q deficiency
References
- Fomin AB, Pastorino AC, Kim CA, et al. "DiGeorge Syndrome: a not so rare disease". Clinics. 65(9). :865-869. (2010)
- Khan K, Wozniak SE, Giannone AL, Abdulmassih ME. "A boy with relentless pruritus: Job’s Syndrome". Am J Case Rep. 17. :104-110. (2016)
- Wissem Hafsi, Talel Badri. "Job Syndrome (Hyperimmunoglobulin E)". StatPearls Publishing. (2019)
- Caragol I, Casanova JL. "Inherited disorders of the Interleukin-12/ Interferon-gamma axis: Mendelian predisposition to mycobacterial disease in man". Inmunología. 22(3). :263-276. (2003)
- Palamaro L, Giardino G, Santamaria F et al. "Interleukin 12 receptor deficiency in a child with recurrent bronchopneumonia and very high IgE levels". Ital J Pediatr. 38(46). (2012)
- Prando C, Samarina A, Bustamante J et al. "Inherited IL-12p40 deficiency". Medicine (Baltimore). 92(2). :109-122. (2013)
- Okada S, Puel A, Casanova J-L, Kobayashi M. "Chronic mucocutaneous candidiasis disease associated with inborn errors of IL-17 immunity". Clin Transl Immunology.. 5(12). :e114. (2016)
- "Omenn syndrome". https://rarediseases.info.nih.gov/diseases/8198/omenn-syndrome/cases/34404#:~:text=Infants%20with%20Omenn%20syndrome%20typically,precede%20the%20onset%20of%20infections.. [2012-03-21]
- Mary Dell Railey, M.D., Yuliya Lokhnygina, et al. "Long Term Clinical Outcome of Patients with Severe Combined Immunodeficiency who Received Related Donor Bone Marrow Transplants without Pre-transplant Chemotherapy or Post-transplant GVHD Prophylaxis". The Journal of Pediatrics. (2009)
- "X-Linked Hyper IgM Syndrome"
- "Dihydrorhodamine (DHR) Flow Cytometric Phorbol Myristate Acetate (PMA) Test, Blood". https://www.mayomedicallaboratories.com/test-catalog/Overview/62765. [2018-01-01]
- Greenberg DE, Goldberg JB, Stock F, et al. "RecurrentBurkholderiaInfection in Patients with Chronic Granulomatous Disease: 11‐Year Experience at a Large Referral Center". Clinical Infectious Diseases. 48(11). :1577-1579. (2009)
- Macedo ACL, Isaac L. "Systemic Lupus Erythematosus and Deficiencies of Early Components of the Complement Classical Pathway". Frontiers in Immunology. 7. (2016)
- Rosain J, Hong E, Fieschi C, et al. "Strains Responsible for Invasive Meningococcal Disease in Patients With Terminal Complement Pathway Deficiencies". J Infect Dis. 215(8). :1331-1338. (2017)
- Van Schaarenburg RA, Magro-Checa C, Bakker JA, et al. "C1q Deficiency and Neuropsychiatric Systemic Lupus Erythematosus". Frontiers in Immunology. 7. (2016)
- "C1q deficiency". https://rarediseases.info.nih.gov/diseases/12958/c1q-deficiency. [2016-03-18]
- Sullivan KE. "Complement Deficiencies". Elsevier. :90-100.e4. (2015). ISBN: 0323298753
- S. Reis E, Falcao DA, Isaac L. "Clinical aspects and molecular basis of primary deficiencies of complement component C3 and its regulatory proteins factor I and factor H". Scand J Immunol. 63(3). :155-168. (2006)
- Wu YL, Hauptmann G, Viguier M, Yu CY. "Molecular basis of complete complement C4 deficiency in two North-African families with systemic lupus erythematosus". Genes & Immunity. 10(5). :433-445. (2009)
- Lintner KE, Wu YL, Yang Y, et al. "Early Components of the Complement Classical Activation Pathway in Human Systemic Autoimmune Diseases". Frontiers in Immunology. 7. (2016)
- "Complement component 2 deficiency". https://medlineplus.gov/genetics/condition/complement-component-2-deficiency/. [2020-08-18]
- Okura Y, Kobayashi I, Yamada M, et al. "Clinical characteristics and genotype-phenotype correlations in C3 deficiency". J Allergy Clin Immunol. 137(2). :640-644.e1. (2015)
- Wang H, Liu M. "Complement C4, Infections, and Autoimmune Diseases". Frontiers in Immunology. 12. (2021)
- "Mannose-binding lectin protein deficiency". https://rarediseases.info.nih.gov/diseases/10309/mannose-binding-lectin-protein-deficiency#ref_11420. [2016-11-23]
- Holdaway J, Deacock S, Williams P, Karim Y. "Mannose-binding lectin deficiency and predisposition to recurrent infection in adults". J Clin Pathol. 69(8). :731-736. (2016)
- "Bruton Agammaglobulinemia"
- Yel L. "Selective IgA deficiency". J Clin Immunol. 30(1). :1-16. (2010)
- "Selective IgA Deficiency". http://www.msdmanuals.com/professional/immunology-allergic-disorders/immunodeficiency-disorders/selective-iga-deficiency. [2016-08-01]
- Cole LA. "False Positive hCG Assays". Elsevier. :229-236. (2010). ISBN: 9780123849076
- "Common Variable Immune Deficiency". https://rarediseases.org/rare-diseases/common-variable-immune-deficiency/. [2017-01-01]
- Sneller MC. "New insights into common variable immunodeficiency". Ann Intern Med. 118(9). :720. (1993)
- Cunningham-Rundles C. "The many faces of common variable immunodeficiency". Hematology Am Soc Hematol Educ Program. 2012. :301-305. (2012)
- Tam JS, Routes JM. "Common Variable Immunodeficiency". American Journal of Rhinology & Allergy. 27(4). :260-265. (2013)
- Agarwal S, Cunningham-Rundles C. "Autoimmunity in common variable immunodeficiency". Ann Allergy Asthma Immunol. 123(5). :454-460. (2019)
- Kliegman RM, Stanton BF, Geme JS, Schor NF, Behrman RE. "Nelson Textbook of pediatrics". Elsevier (2011). (2011). ISBN: 9781437707557
- A Wesley Burks, Stephen Holgate, Robyn O'Hehir, et al. "Middleton's Allergy: Principles and Practice". Elsevier. (2019). ISBN: 9780323546980