Summary
Cyanotic heart defects (CHDs) are congenital cardiac malformations that commonly affect the atrial walls, e.g., the right atrium (RA) or left atrium (LA); ventricular walls, e.g., the left ventricle (LV) or right ventricle (RV); heart valves; or large blood vessels. Common causes include genetic defects (e.g., DiGeorge syndrome), maternal conditions (e.g., diabetes), and spontaneous genetic mutations. Pathophysiologically, cyanotic heart defects are characterized by a right-to-left cardiac shunt, which leads to deoxygenated blood entering the systemic circulation. The resulting hypoxemia manifests clinically as cyanosis, which may occur as acute, life-threatening episodes. Other symptoms include failure to thrive, characteristic heart murmurs, and symptoms of heart failure. Diagnosis is confirmed through visualization of the defect on echocardiography. Further diagnostic findings include low oxygen saturation and characteristic x-ray findings. Heart defects requiring treatment are repaired via catheter procedures or surgery. Supportive medical therapy is required in patients with heart failure (e.g., diuretics, inotropic agents) or if surgery cannot be performed (e.g., prostaglandin). If untreated, most cyanotic heart defects are fatal within the first year of life.
Overview of cyanotic congenital heart defects
Commonly associated conditions and risk factors of cyanotic congenital heart defects
| Most common associated conditions and risk factors | |
|---|---|
| Tetralogy of Fallot |
|
| Transposition of great vessels |
|
| Tricuspid valve atresia |
|
| Ebstein anomaly |
|
| Total anomalous pulmonary venous return |
|
| Persistent truncus arteriosus |
|
| Hypoplastic left heart syndrome |
|
General pathophysiological processes
- CHDs result from the disruption of the normal cardiac morphogenesis sequence.
- CHDs may lead to the formation of connections between the right and left heart (i.e., shunts).
- In cyanotic CHDs: right-to-left cardiac shunting → blood flow from the right to the left heart → deoxygenated blood entering the systemic circulation → cyanosis

General clinical features
- “Blue babies”: pale gray or blue skin color caused by cyanosis
- Feeding problems and failure to thrive
- Exertional dyspnea, tachypnea, and fatigue
- Hypoxemia [1][2]
- Symptoms of heart failure (see “Acyanotic congenital heart defects”)
- Nail clubbing may occur later in life.
In approximately 50% of infants with CHDs, a routine newborn physical examination reveals no abnormalities. A normal physical examination does not rule out CHD. [3][4]

General treatment considerations [5]
-
Ductal-dependent CHDs: a group of CHDs that require the patent ductus arteriosus (PDA), which supplies either pulmonary or systemic circulation, to sustain life until surgery can be performed [6]
-
PDA supplies systemic circulation in the following:
- Transposition of the great arteries
- Hypoplastic left heart syndrome
- Coarctation of the aorta
- Critical aortic valve stenosis
-
PDA supplies pulmonary circulation in the following:
- Tricuspid atresia
- Critical pulmonary valve stenosis, tetralogy of Fallot
-
Treatment: administration of prostaglandin E1 infusion
- Example: IV alprostadil infusion (off-label) [7][8]
- Mechanism of action: prevents the ductus arteriosus from closing, creating an intentional shunt to allow mixing of deoxygenated with oxygenated blood
- Monitor respiratory status and hemodynamic parameters. [7]
-
PDA supplies systemic circulation in the following:
- In patients with heart failure [9]
- Decrease fluid volume and lower pulmonary vascular resistance with diuretics or ACE inhibitors.
- Improve the contractility of the heart with inotropic agents (e.g., digoxin).
-
Antibiotic prophylaxis for bacterial endocarditis during dental procedures [10][11]
- For all patients with unrepaired cyanotic CHDs
- During the first six months after surgery
- Surgical repair is required in most cases.
Urgently consult critical care and pediatric cardiology for any neonate or infant presenting with cyanosis or shock. [7]
The “5 Ts” of cyanotic CHDs: Tetralogy of Fallot, Transposition of the great vessels, Tricuspid valve anomalies, Total anomalous pulmonary venous return, and persistent Truncus arteriosus
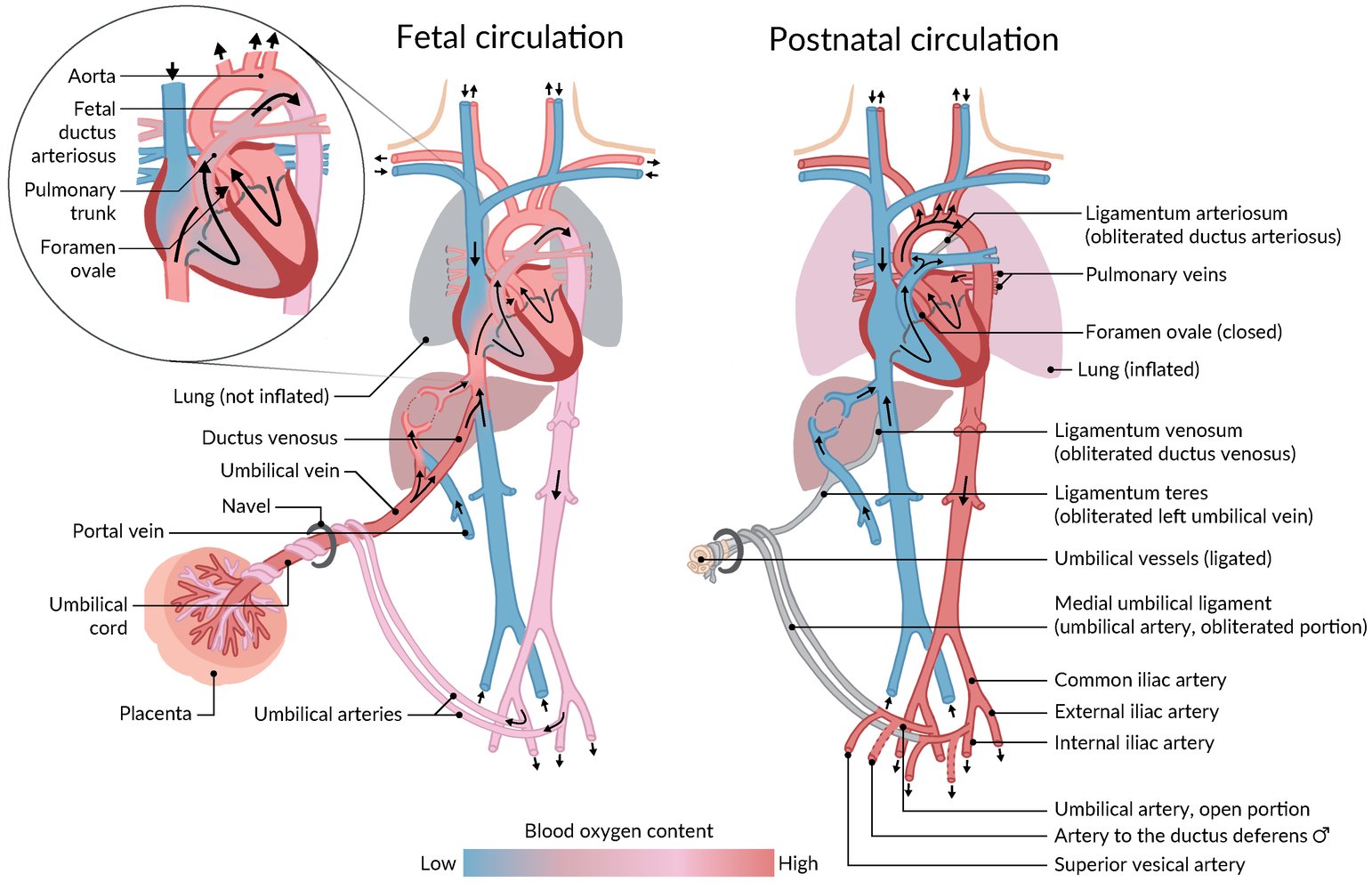
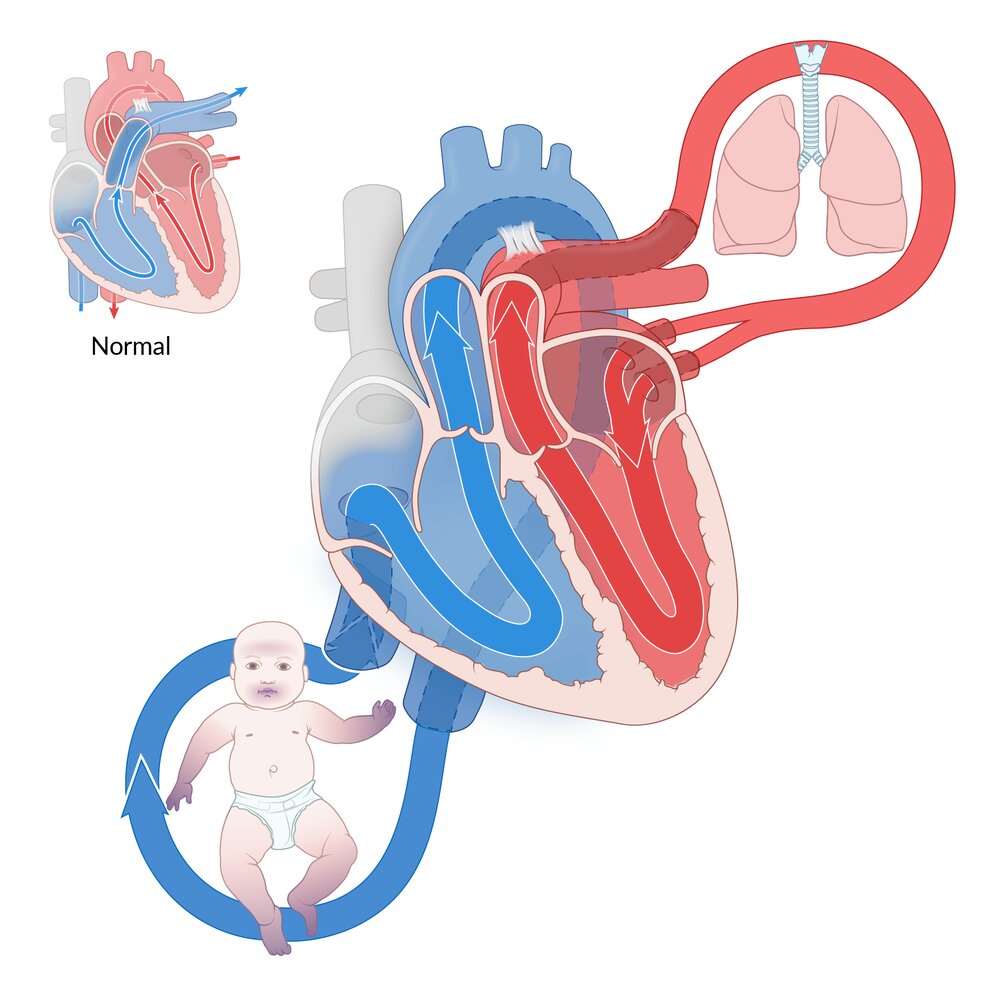
Fetal circulation (left) is connected to the maternal circulation via the placenta. The umbilical vein delivers oxygen-rich blood from the placenta to the fetus, and the umbilical arteries return the blood to the placenta for oxygenation. Blood bypasses pulmonary circulation entering from the right to the left atrium via the foramen ovale. Deoxygenated blood entering the right atrium through the superior vena cava bypasses pulmonary circulation via the ductus arteriosus, which connects the pulmonary trunk to the aorta.
Postnatal circulation (right) is formed under the influence of postnatal pulmonary ventilation and the disconnection of umbilical vessels. These alterations cause a change in pressure within the circulatory system, leading to the opening of the pulmonary circulation and obliteration of the umbilical vessels and bypass structures.
Blue: deoxygenated blood; Red: oxygenated blood; Pink: mixed blood
© AMBOSS
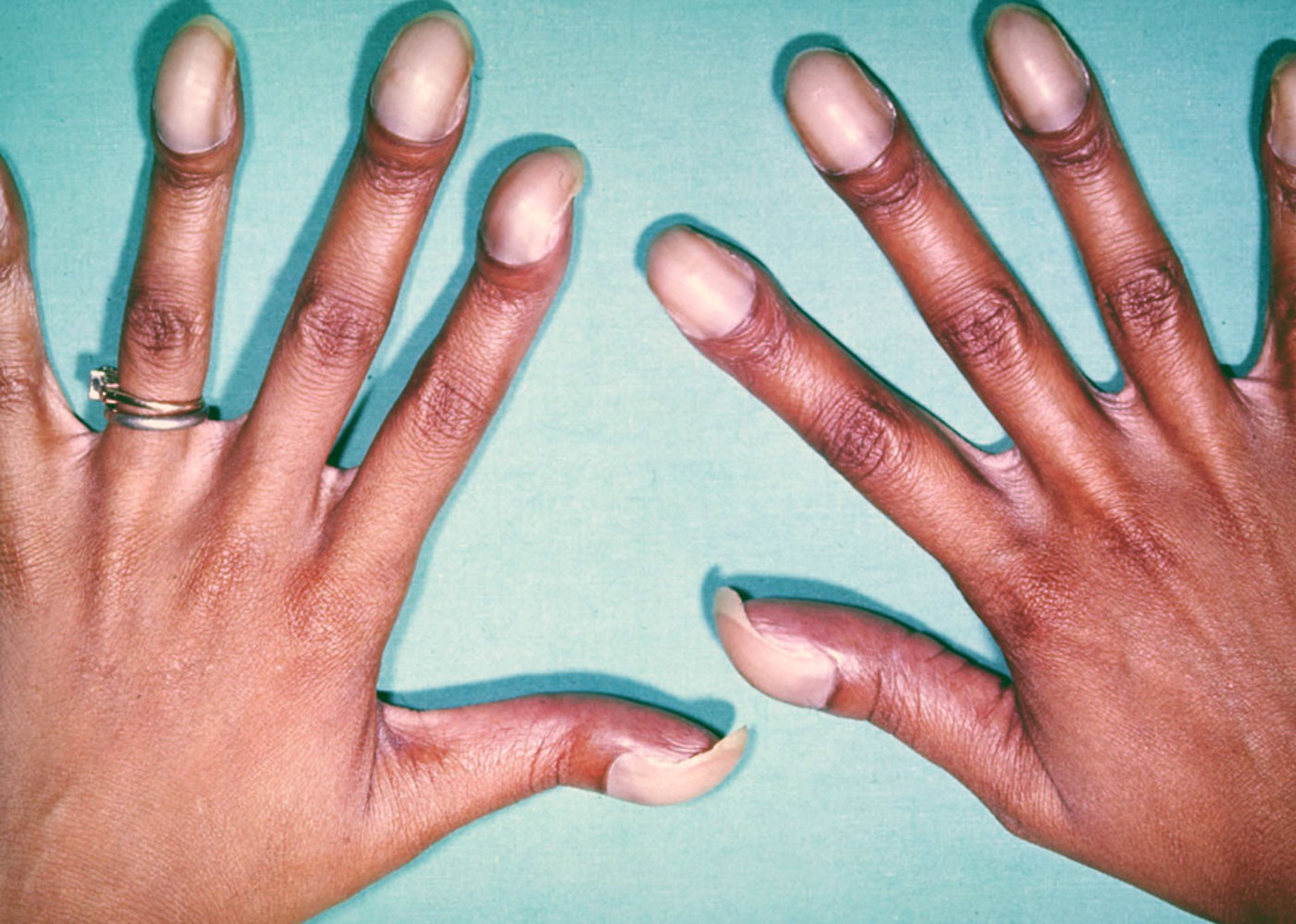
There is bulbous enlargement of the distal phalanges and the nail curvatures are markedly increased.
Source: “CongenitalHeartCase-133” by Herbert L. Fred, MD and Hendrik A. van Dijk, openstax cnx, licensed under CC BY-SA 3.0.
Tetralogy of Fallot
Overview [12]
-
Definition: tetralogy of Fallot (TOF) is the simultaneous occurrence of the following four defects:
- Right ventricular outflow tract obstruction (RVOTO) due to pulmonary infundibular stenosis
- Right ventricular hypertrophy (RVH)
- Ventricular septal defect (VSD)
- Overriding aorta (the aorta is displaced above the VSD)
-
Other cardiac defects associated with TOF [13]
- Atrial septal defect (ASD)
- PDA
- Anomalous coronary arteries [14]
RVOTO: Right ventricular hypertrophy, Ventricular septal defect, Overriding aorta are the characteristics of Tetralogy of FallOt.
Epidemiology
- Prevalence: 4–5/10,000 live births in the US [15]
- The most common cyanotic CHD [16]
Etiology
- Typically sporadic [17]
- Associated with the following genetic disorders:
- DiGeorge syndrome [18][19]
- Down syndrome [20][21]
- Maternal exposures during pregnancy
- Alcohol consumption
- Phenylketonuria [22][23]
- Diabetes [24][25]
Pathophysiology
- During fetal development, anterior and superior deviation of the infundibular septum → misaligned VSD with overriding aortic root and subsequent RVOTO
-
Physiologic blood flow is determined by the severity of RVOTO.
- A large VSD → equal pressures in the right and left ventricles → blood flow along the path of least resistance
- Severe RVOTO → flow from RV to LV → desaturated blood entering the circulation via the aorta

Clinical findings
- Infants with mild TOF may be asymptomatic on initial examination.
-
Mild to severe cyanosis, which depends on the severity of the RVOTO
- Mild obstruction → more pronounced left-to-right shunt via VSD → little or mild cyanosis
- Severe obstruction → more pronounced right-to-left shunt via VSD → severe cyanosis
-
Tet spells: intermittent hypercyanotic, hypoxic episodes with a peak incidence at 2–4 months after birth ; [26][27]
- Associated with psychological and physical stress (e.g., crying, feeding, defecation)
- Caused by either an increase in pulmonary vascular resistance (PVR) or a decrease in systemic vascular resistance (SVR) → intermittent worsening of RVOTO and ↑ right-to-left shunting [28][29]
- Spells occur suddenly and are potentially lethal.
- Untreated children tend to squat: squatting → ↑ SVR → ↓ right-to-left shunt → ↑ blood flow to pulmonary circulation → ↓ hypoxemia → relief of symptoms
-
Cardiac examination findings
- Harsh systolic ejection murmur at the left upper sternal border
- Single S2
- Possible RV heave and systolic thrill
- CHF symptoms (see “Overview” in “Heart failure”)
In patients with TOF, the auscultated murmur is determined by the amount of blood flow across the RVOTO. Therefore, during tet spells, the murmur may disappear.

Diagnostics
- Prenatal diagnosis: Most cases are now diagnosed before birth due to improvements in fetal echocardiography. [30]
-
Imaging
-
Echocardiography (confirmatory test)
- Detection of the main features of TOF
- Quantification of right ventricular outflow tract pressure gradient
- Supplementary cardiac catheterization may be performed.
-
Chest x-ray
- Boot-shaped heart (due to upturned cardiac apex and RVH)
- Normal or decreased pulmonary vascular markings
- Concave pulmonary artery segment
-
Echocardiography (confirmatory test)
-
ECG
- Right axis deviation
- Prominent anterior R waves (V1–V2)
- Prominent posterior S waves
- Right atrial enlargement and RVH (P pulmonale)
- Pulse oximetry: ↓ SpO2
- Hyperoxia test: helps to distinguish cardiac from pulmonary causes of cyanosis


Treatment [5][31]
- Severe RVOTO: PGE1 infusion until surgery [8]
-
Acute hypoxia (tet spells) ; [7] [32]
-
Initial interventions
- Administer oxygen; (e.g., 100% FiO2 via NRB).
- Knee to chest position, squatting
- IM morphine (off-label) [7]
-
Subsequent interventions: Consider if no improvement.
- IV sodium bicarbonate (off-label) to correct metabolic acidosis [7]
- IV fluid resuscitation
- Vasopressors, e.g., phenylephrine (off-label) [7]
- IV beta blockers, e.g., propranolol (off label) [7]
-
Initial interventions
-
Heart failure
- Inotropic agents (e.g., digoxin) and loop diuretics (e.g., furosemide)
- ACE inhibitors are not recommended in patients with TOF because they may decrease SVR and promote tet spells.
-
Surgical repair: performed within the first year of life ; [33][34]
- VSD repair: patch closure of the ventricular septal defect ensuring correct aortal positioning above the left ventricle
- Enlargement of the RVOTO: resection of the obstructive infundibular musculature
-
If early surgical management is not possible: palliative shunts ; [35] [36]
- Blalock–Thomas–Taussig shunt: a palliative surgical procedure for TOF that connects the subclavian artery to the pulmonary artery with an interposed graft
- Central shunt
- Others (e.g., Potts shunt, Cooley shunt, Waterston shunt, Sano shunt)
- Follow-up care: to prevent long-term complications, such as heart failure, arrhythmias (e.g., ventricular tachycardia), and neurodevelopmental impairment
Prognosis
- Without surgery, ∼ 50% of patients do not live past the first three years of life. [37]
- With corrective surgery, over 90% of patients live to > 25 years of age. [38]
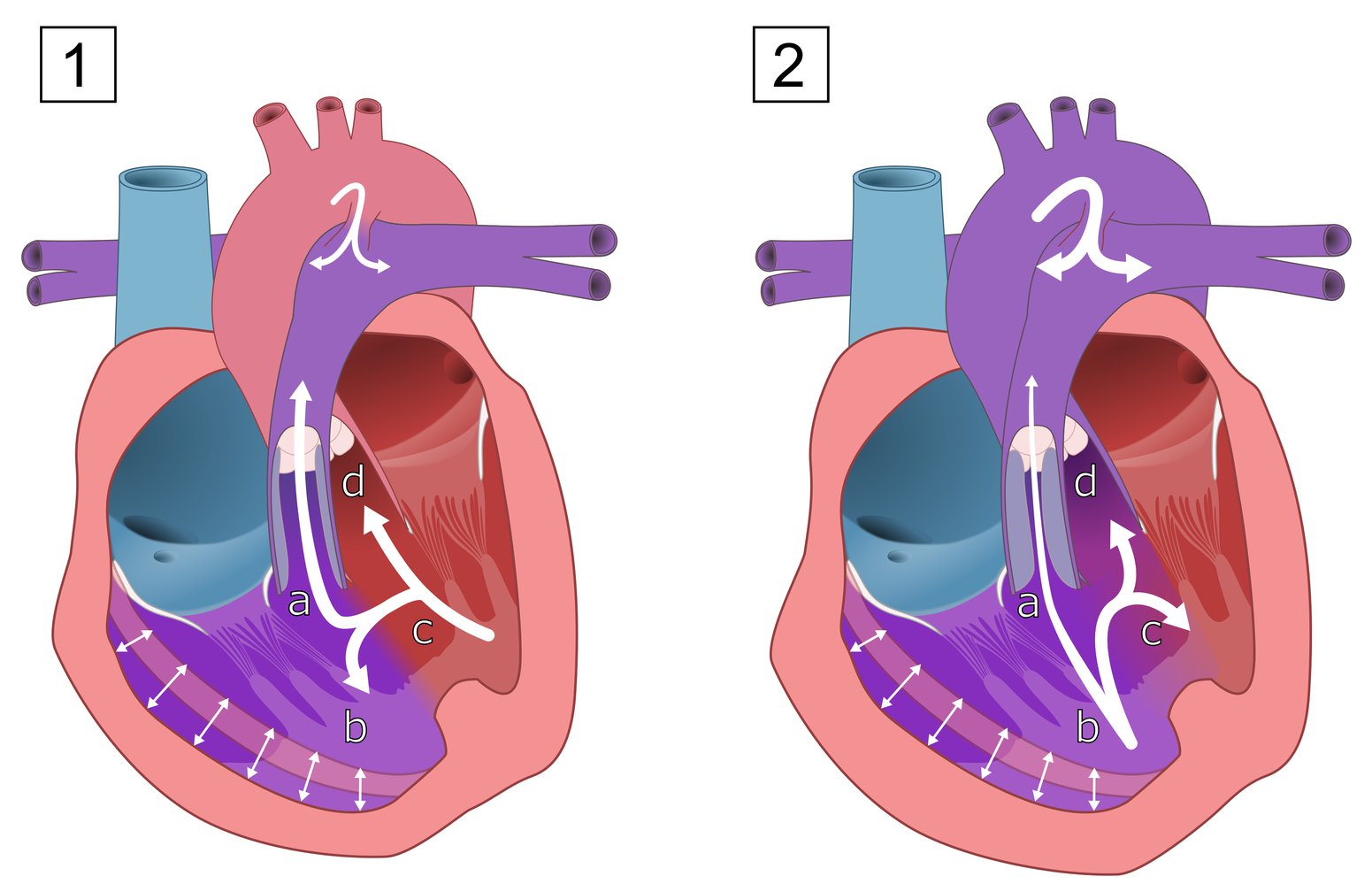
Tetralogy of Fallot is the simultaneous occurrence of:
(a) Pulmonary stenosis
(b) Right ventricular hypertrophy
(c) Ventricular septal defect (VSD)
(d) An overriding aorta (above the VSD)
The hemodynamic effects depend on the extent of right ventricular outflow tract obstruction. With mild obstruction (1), a left-to-right shunt is more pronounced and there is mild cyanosis. With severe obstruction (2), a right-to-left shunt is more pronounced, which results in severe cyanosis. Pulmonary circulation may be supplied via a patent ductus arteriosus.
© AMBOSS
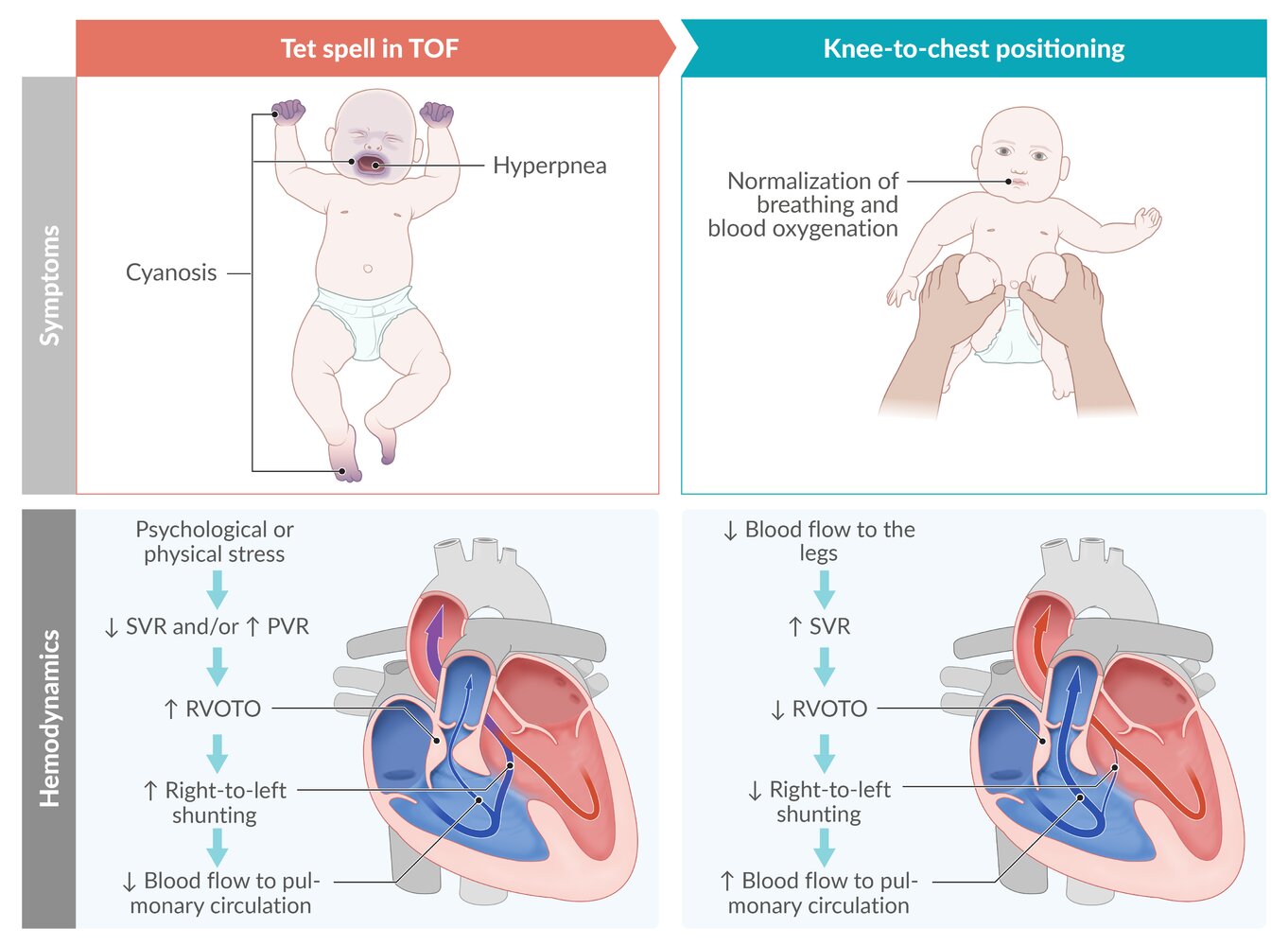
Tet spells (hypercyanotic spells) are caused by a sudden decrease in systemic vascular resistance (SVR) or increase in pulmonary vascular resistance (PVR), leading to intermittent worsening of right ventricular outflow tract obstruction (RVOTO) and increased right-to-left shunting. The resulting decrease in blood flow to the pulmonary circulation results in decreased blood oxygenation.
Knee-to-chest positioning or, in older children, squatting relieve symptoms by increasing SVR, which in turn reduces right-to-left-shunting and improves blood flow to the lungs.
© AMBOSS
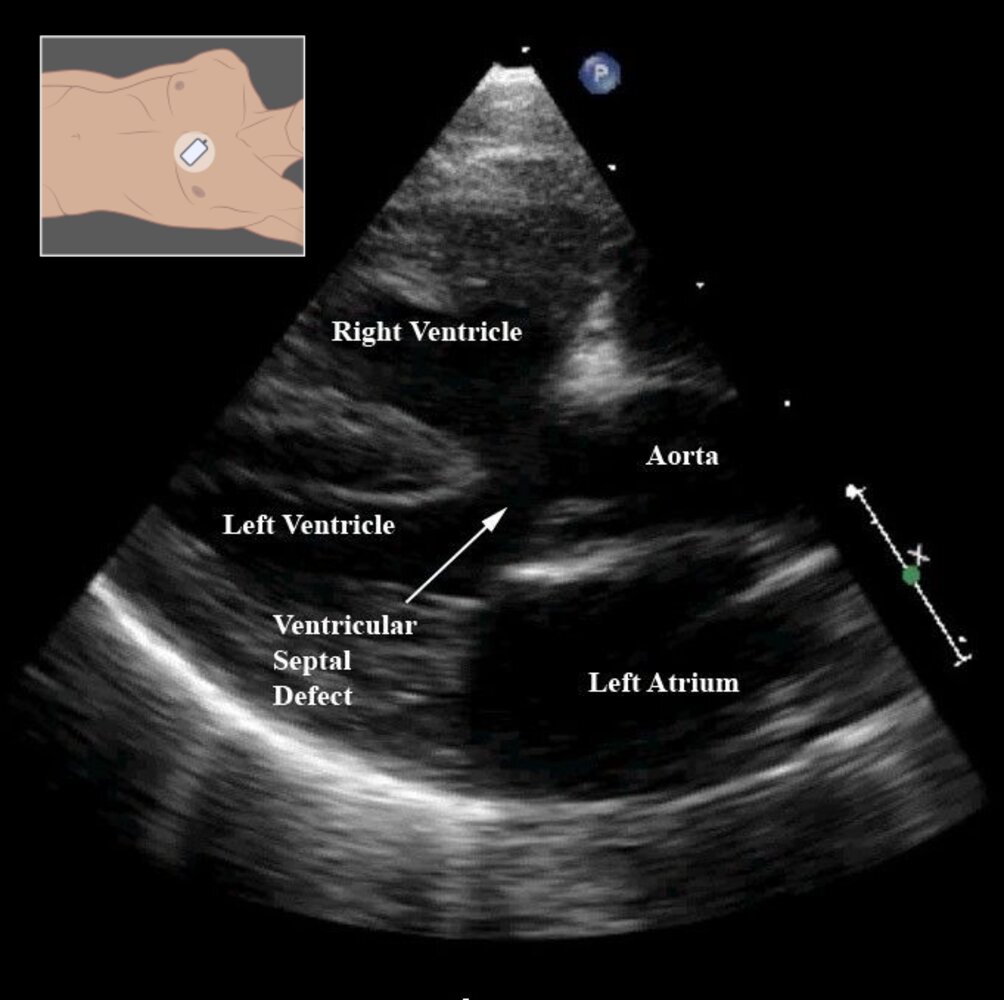
Echocardiography (transthoracic; modified parasternal long axis view)
The ascending aorta over rides a large ventricular septal defect. The right ventricular wall is notably thickened from hypertrophy.
These are characteristic findings in tetralogy of Fallot.
Source: “Figure 8- in: Tetralogy of Fallot” by Frederique Bailliard & Robert H Anderson, Orphanet Journal of Rare Diseases, licensed under CC BY 2.0. Modifications: - added illustration.
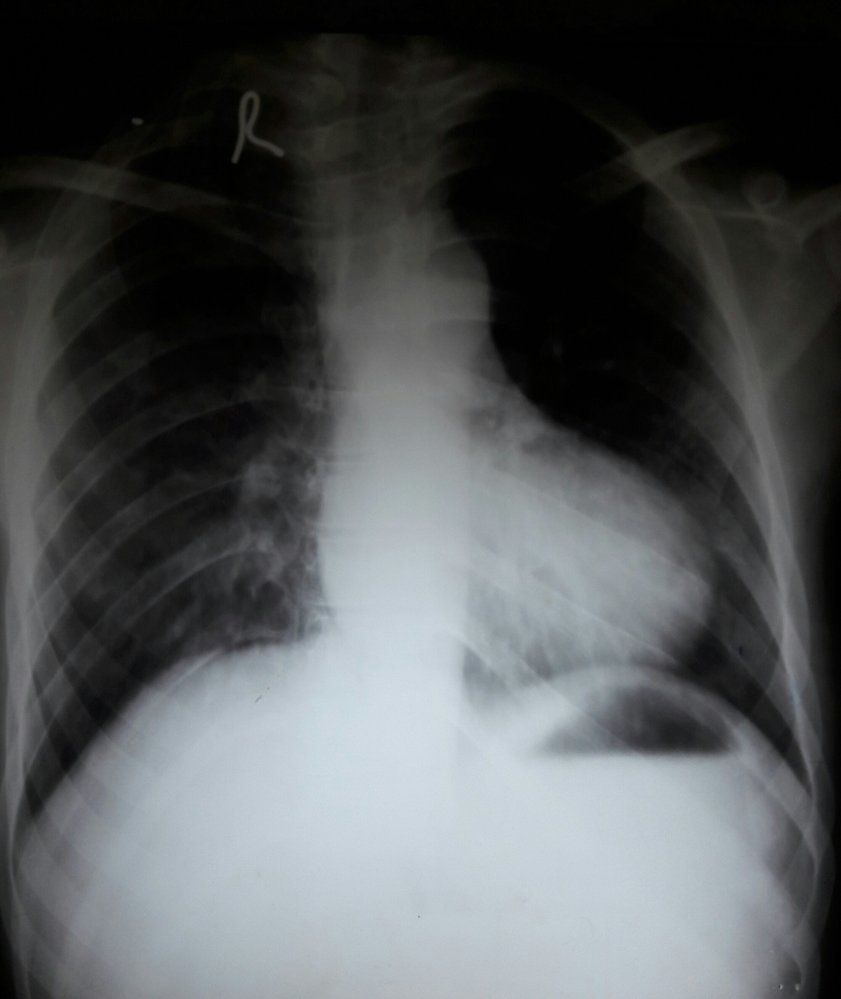
X-ray chest (PA view) of a patient with a history of tetralogy of Fallot
The cardiac apex is upturned from right ventricular hypertrophy, and the pulmonary artery segment is small and concave in appearance as a result of pulmonary stenosis. These findings produce a characteristic boot-shaped heart (green overlay; cf. illustration), seen in some patients with tetralogy of Fallot. Pulmonary vascular markings are diminished as a result of reduced blood flow to the lungs.
White dashed outline: normal size and shape of the heart
Source: “Boot-shaped heart” by Medicalpal, Wikimedia Commons, licensed under CC BY-SA 4.0. The supplementary image with overlays of relevant areas was adapted from the image mentioned above and licensed under CC BY-SA 4.0.
{kind=link}
Transposition of the great vessels (TGV)
Overview
- Definition: anatomical reversal of the aorta and the pulmonary artery
-
Other cardiac defects associated with TGV
- VSD [39]
- Left ventricular outlet obstruction [40]
- Abnormal valves and/or coronary arteries
Epidemiology
- Prevalence: 2–4/10,000 live births in the US [15][16]
- Accounts for ∼ 20% of all cyanotic CHD cases [16]
Etiology
- Often multifactorial or unknown
- Intrauterine risk factors: infants born to mothers with diabetes [5][24]
- Genetic syndromes: seen in ∼ 1% of patients with DiGeorge syndrome [41]
Pathophysiology [42]
- Failed spiraling of the aorticopulmonary septum → RV emptying into the aorta and LV into the pulmonary artery → complete isolation of the pulmonary and systemic circuits → ↓ oxygenated blood entering the systemic circulation
- Fatal, unless mixing occurs via an intracardiac shunt (e.g., PFO, VSD, ASD) or via an extracardiac connection (e.g., PDA)
")


Clinical findings
- Postnatal cyanosis (not affected by exertion or supplemental oxygen)
- Tachypnea
-
Cardiac examination findings
- Single, loud S2
- There is often no murmur.
- Diminished femoral pulses
- If concurrent VSD is present: systolic murmur at the left sternal border
Diagnostics [5]
- Prenatal diagnosis: TGV is difficult to diagnose with fetal ultrasound. [43]
-
Imaging
-
Echocardiography (confirmatory test)
- Pulmonary artery arising from the left ventricle
- Aorta arising from the right ventricle
-
Chest x-ray
-
“Egg on a string” appearance of the heart
- The enlarged, globular heart resembles an egg lying on its side.
- The string represents the superior mediastinum that appears narrow due to stress-induced thymic atrophy.
- ↑ Pulmonary vascular markings
-
“Egg on a string” appearance of the heart
- MRI (rarely done) showing the aorta arising from the right ventricle and the pulmonary trunk originating from the left ventricle
-
Echocardiography (confirmatory test)
- ECG: often normal
- Pulse oximetry: ↓ SpO2

Treatment [44]
-
Initial postnatal management: Initiate mixing between the two parallel circulations to ensure adequate systemic oxygenation.
- Infusion of PGE1 to prevent closure of the PDA [45]
-
Balloon atrial septostomy [46][47]
- Objective: to enhance atrial mixing if PGE1 administration is insufficient, and to alleviate hypoxemia [48]
- Procedure: right heart catheterization with the creation or enlargement of an existing ASD
-
Surgical repair: recommended within the first two weeks of life [49][50]
- Arterial switch procedure: reversal of the aorta and pulmonary artery with insertion into the anatomically correct ventricle as well as correction of coronary artery supply [51][52]
-
Rastelli procedure [53][54]
- Indication: TGV with concurrent large VSD and LVOTO repair
- Procedure: creation of a conduit from the LVOTO through the VSD to the aorta and creation of a conduit from the RV to the pulmonary artery

Prognosis
- Without treatment, 90% of patients with TGA die within the first year of life. [55]
- Long-term survival beyond 15–20 years of age is ∼ 90%. [51]
TGV is a cyanotic congenital heart disease in which the pulmonary artery arises from the left ventricle and the aorta from the right ventricle, resulting in the formation of parallel circulatory systems. The left ventricle pumps oxygenated blood into the pulmonary circulation (red) and the right ventricle pumps deoxygenated blood into the aorta (blue), resulting in cyanosis.
TGV is fatal unless a shunt between the two circulations is present (i.e., a patent ductus arteriosus or a patent foramen ovale) or a surgical intervention is performed in the neonatal period.
© AMBOSS
In transposition of the great vessels, deoxygenated blood enters the systemic circulation. Survival after birth is only possible via an intracardiac shunt (e.g., across a patent ductus arteriosus and/or a patent foramen ovale). The shunt diverts oxygenated blood from the pulmonary circulation into the systemic circulation (white arrows).
© AMBOSS
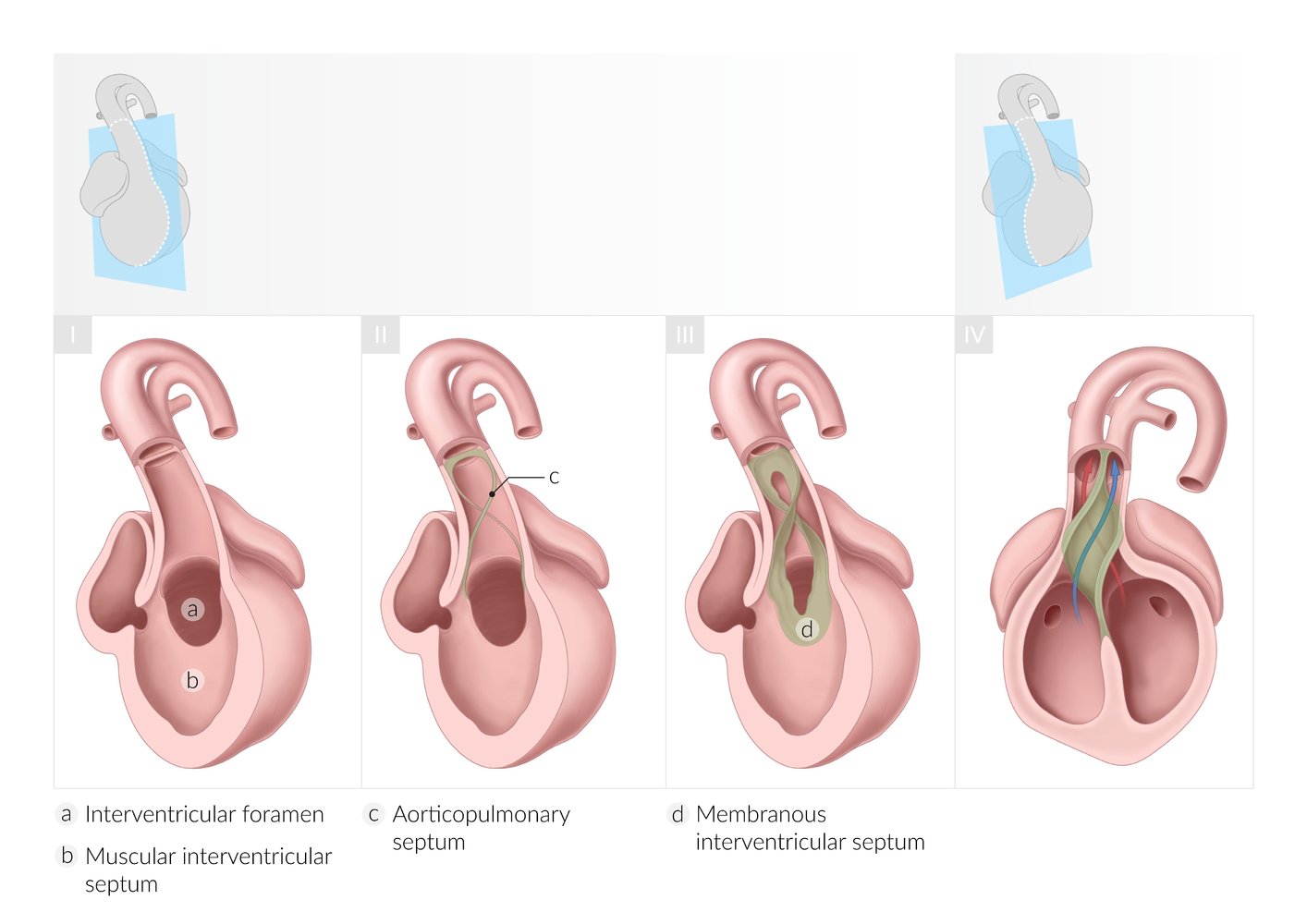
I: Development of the muscular interventricular septum and interventricular foramen
- The primary ventricle is a single cavity that is separated from the atrial cavity by the endocardial cushion. Ventricular septation begins with the formation of a muscular interventricular septum, which arises from the floor of the primitive ventricle and grows cranially toward the endocardial cushion. The interventricular foramen is the space between the endocardial cushion and the growing edge of the muscular interventricular septum and it allows for interventricular communication.
II: Development of the aorticopulmonary septum
- The aorticopulmonary septum develops from ridges of tissue derived from neural crest cells within the ventricular outflow tract (truncus arteriosus). These ridges are continuous with the bulbar ridges of the bulbar cordis (not shown here).
III: Development of the membranous interventricular septum and closure of the interventricular foramen
- The membranous interventricular septum develops from the bulbar ridges and the endocardial cushion.
- The caudal end of the aorticopulmonary septum fuses with the muscular interventricular septum and closes the interventricular foramen.
IV: Division of the ventricular outflow tract
- The aorticopulmonary septum fuses and rotates 180°, dividing the outflow tract into the aorta (from the left ventricle) and pulmonary trunk (from the right ventricle).
© AMBOSS
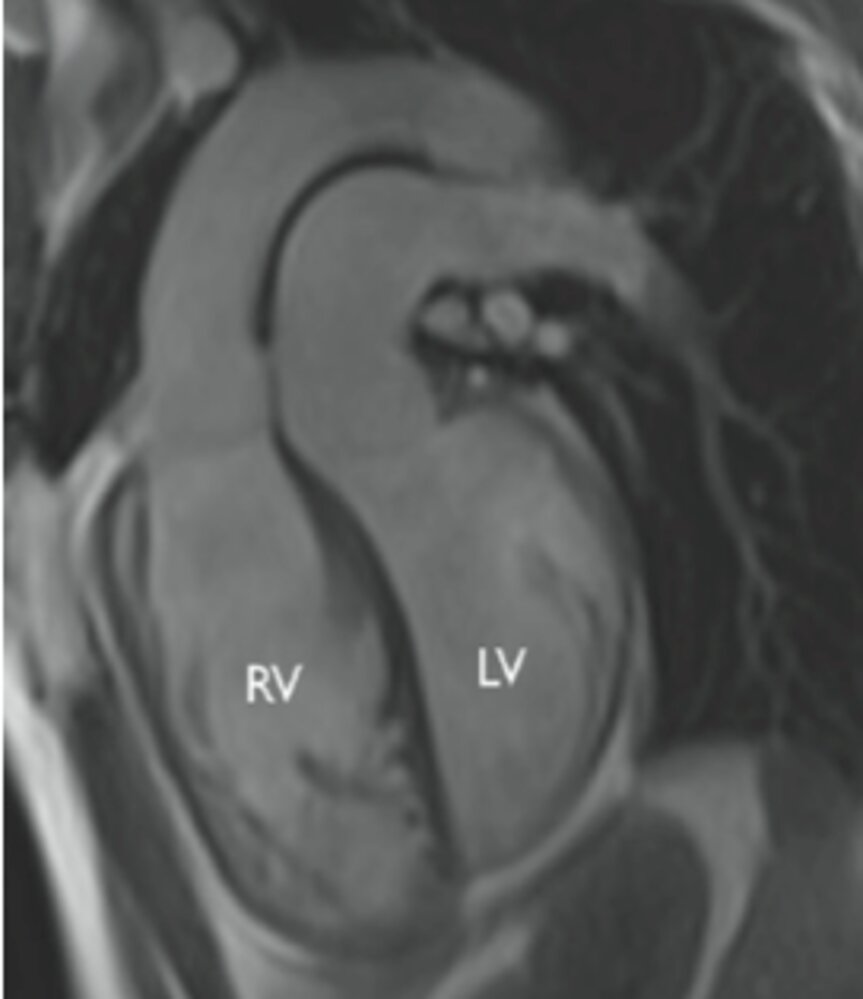
MRI cardiac (oblique sagittal plane)
The aorta (Ao) arises from the right ventricle (RV), while the pulmonary trunk (pulmonary artery; PA) originates from the left ventricle (LV).
Source: “Figure 5A. in: The Role of Cardiovascular Magnetic Resonance in Pediatric Congenital Heart Disease” by Hopewell N Ntsinjana, Marina L Hughes & Andrew M Taylor, Journal of Cardiovascular Magnetic Resonance, BioMed Central, licensed under CC BY 2.0. Modifications: Removed "A.". The supplementary image with overlays of relevant areas was adapted from the image mentioned above (© AMBOSS).
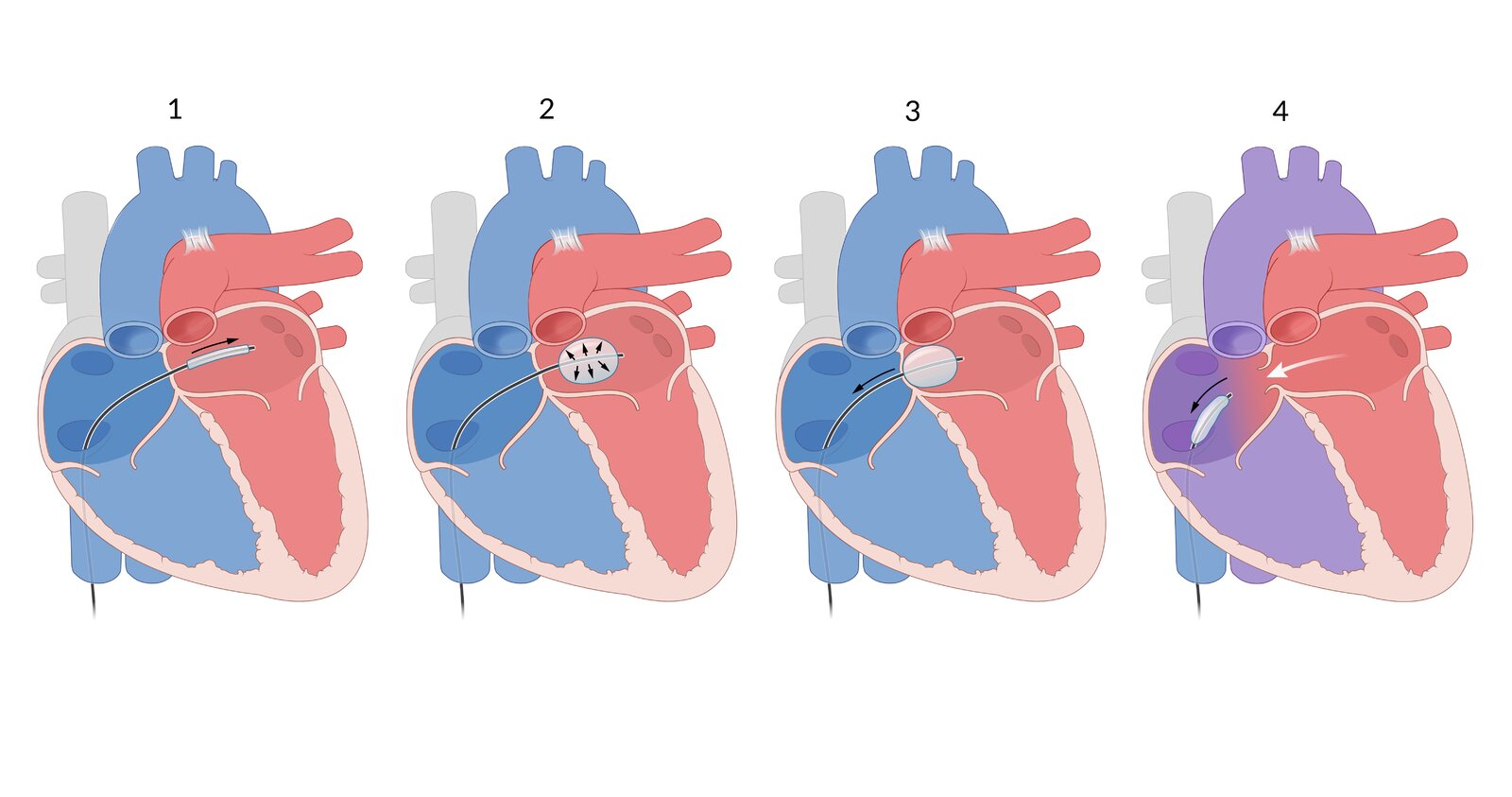
The heart with transposition of the great vessels (TGV)
Slight oxygen saturation of the blood occurs via the ductus arteriosus as well as a small atrial septal defect. This left-to-right shunt is enlarged using balloon atrial septostomy:
(1) A balloon catheter is advanced into the right atrium through the inferior vena cava, then through the patent foramen ovale into the left atrium.
(2) The correct positioning of the balloon is controlled via ultrasound. The balloon is expanded with a saline solution.
(3) The balloon is pulled back through the foramen ovale into the right atrium, resulting in the fossa ovalis of the atrial septum tearing and creating an enlarged left-to-right shunt.
(4) This results in an increase of oxygenated blood in the systemic circulation and a corresponding rise in saturation, usually directly following the procedure.
However, balloon atrial septostomy is only a temporary measure. Patients with TGV will undergo an arterial switch surgery, which will result in a normal circulation of blood in the body.
© AMBOSS
Tricuspid valve atresia
Overview
-
Definition
- Absent or rudimentary tricuspid valve resulting in no blood flow between the RA and the RV
- Patient survival is only possible if there are interatrial and interventricular communications (see “Other cardiac defects” below). [56]
-
Other cardiac defects associated with tricuspid valve atresia
- ASD
- VSD
- RV hypoplasia
- Other defects (e.g., pulmonary outflow obstruction, aortic stenosis, aortic coarctation)
Epidemiology
- Prevalence: 0.2–1.2/10,000 live births in the US [16]
- Third most common cyanotic CHD
Pathophysiology
- Tricuspid atresia is accompanied by RV hypoplasia and RA dilation due to volume overload (univentricular heart).
-
Circulation depends on the presence of interatrial and interventricular communications.
- Blood in the RA flows through the ASD into the LA → venous blood mixes with arterial blood in the left heart → cyanosis
- Blood can only reach the RV and pulmonary system via a VSD.

Clinical features
- Central cyanosis (occurs within days after birth)
- Tachypnea
-
Cardiac examination findings
- Holosystolic murmur at the lower left sternal border
- Single S2
- Jugular venous distention with a prominent A wave
- Diminished peripheral pulses
Diagnostics
- Prenatal diagnosis: fetal echocardiography at 18–22 weeks' gestation [57]
-
Imaging
-
Echocardiography (confirmatory test)
- Absent tricuspid valve
- ASD
- RV hypoplasia
-
Chest x-ray
- Mild cardiomegaly
- Abnormal pulmonary markings
-
Echocardiography (confirmatory test)
-
ECG [58]
- LVH with left axis deviation
- Tall P waves
- Minimal R waves in precordial leads
- Pulse oximetry: ↓ SpO2
Treatment
-
Initial postnatal management: cardiopulmonary stabilization until surgical palliation
- PGE1 infusion
- Cardiorespiratory support (e.g., inotropes, mechanical ventilation, supplemental oxygen)
- Surgical repair: three-stage surgical palliation to isolate both pulmonary and systemic circulations (see “Surgical repair” in “Hypoplastic left heart syndrome”)
- Optional temporary measures
- In cases with concurrent pulmonary valve atresia or severe stenosis: systemic-to-pulmonary shunt (Blalock-Taussig shunt) to improve perfusion
- Risk of pulmonary hypertension: pulmonary artery banding to control pulmonary hyperperfusion
Prognosis
- With surgery, ∼ 90% of patients live to the age of one year and the 10-year survival rate is ∼ 80%. [56]
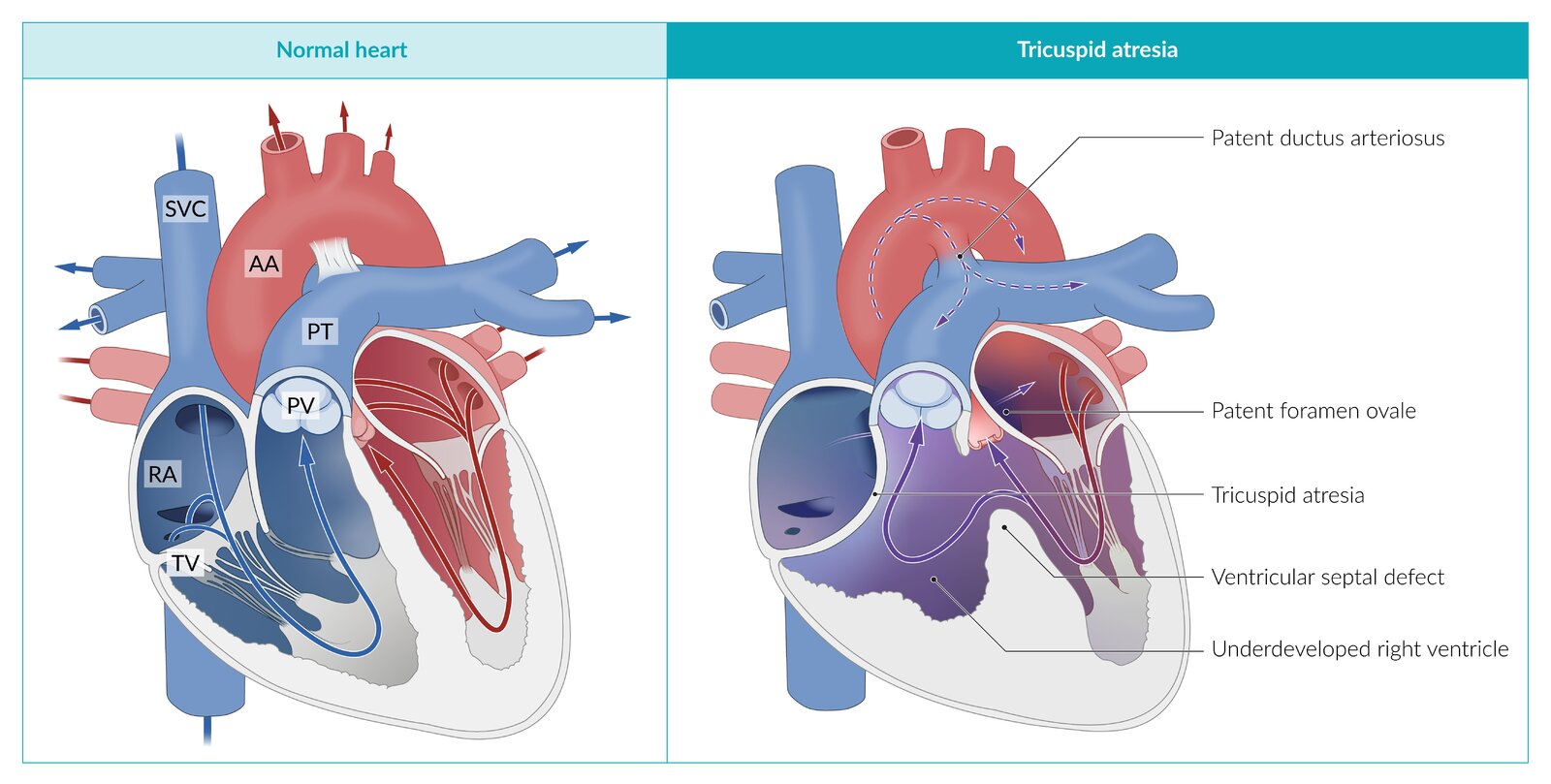
AA: ascending aorta
PT: pulmonary trunk
PV: pulmonary valve
RA: right atrium
SVC: superior vena cava
TV: tricuspid valve
© AMBOSS
Ebstein anomaly
Overview
- Definition: malformed tricuspid valve leaflets that are displaced into the right ventricle with subsequent tricuspid valve regurgitation and right heart enlargement (known as RV “atrialization”)
-
Other cardiac defects associated with Ebstein anomaly
- Interatrial communication (e.g., PFO, ASD): seen in ∼ 90% of patients [59]
- Conduction disorders (e.g., Wolff-Parkinson-White syndrome, supraventricular tachycardia) [60]
Epidemiology
- Prevalence: 0.5–1/10,000 live births in the US [61]
- Accounts for < 1% of all CHD cases [16]
Etiology
- Multifactorial (most cases are sporadic) [62]
- Associated with prenatal lithium exposure [63][64]
- Isolated genetic defects (e.g., MYH7 which encodes β-myosin heavy chain)
Pathophysiology [59]
-
Incomplete delamination (i.e., separation of valve tissue from the myocardium) during valve development can lead to one or more of the following:
- Incomplete closure of the right atrioventricular valve and reduction in RV volume → tricuspid regurgitation→ RA dilation → right heart failure
- RV atrialization → reduced function of the RV → insufficient systolic contraction → functional pulmonary valve atresia → ↓ pulmonary flow → ↓ cardiac output → systemic hypoperfusion, dyspnea, and fatigue
- Bulging of the large sail-like anterior tricuspid leaflet into the RV → RVOTO → ↑ right heart pressure → ↑ flow through the patent foramen ovale → right-to-left shunt → cyanosis


Clinical findings [59]
-
The clinical presentation of Ebstein anomaly varies according to the severity of the abnormality. [65]
- Mild apical displacement → asymptomatic presentation throughout adulthood
- Moderate tricuspid leaflet displacement → cyanosis or heart failure in infancy or childhood
- Severe displacement → in utero heart failure → nonimmune hydrops fetalis → death
-
Neonates and children
- Cyanosis
- Severe cardiomegaly and heart failure
- Respiratory distress
-
Adolescents and adults
- Arrhythmias e.g., Wolff-Parkinson-White syndrome
- Exertional dyspnea and fatigue
- Palpitations and sudden cardiac death
-
Cardiac examination findings
- Loud S1
- Widely split S1 and S2
- Holosystolic murmur at the left sternal border
Diagnostics
- Prenatal diagnosis: fetal echocardiography
-
Imaging
-
Echocardiography (confirmatory test)
- Enlarged right heart
- Tricuspid regurgitation
-
Chest x-ray
- Mild to severe cardiomegaly
- RA enlargement
- MRI : apical displacement of the septal and posterior tricuspid leaflets
-
Echocardiography (confirmatory test)
-
ECG
- Tall and broad P waves
- First-degree atrioventricular block
- Right axis deviation
- Right bundle branch block
- Findings specific for WPW syndrome (e.g., delta wave)
- Pulse oximetry: ↓ SpO2

Treatment
-
Initial postnatal management: stabilization of cardiorespiratory status until PVR decreases and surgery can be performed
- PGE1 infusion
- Inhaled nitric oxide or infusion of milrinone
-
Surgical repair
- Objective: creation of a systemic-pulmonary shunt, repair of the tricuspid valve, and reconstruction of the right ventricle
- Age-related considerations
- Neonates and infants: should be avoided in infancy and delayed as long as possible [66]
- Children and adults: recommended if concomitant heart failure or progressive RV dysfunction is present [67]
- Arrhythmias: catheter ablation of the aberrant pathway [68]
Prognosis
- Varies according to the severity of disease
- Around 30% of patients die within the first year of life. [65]
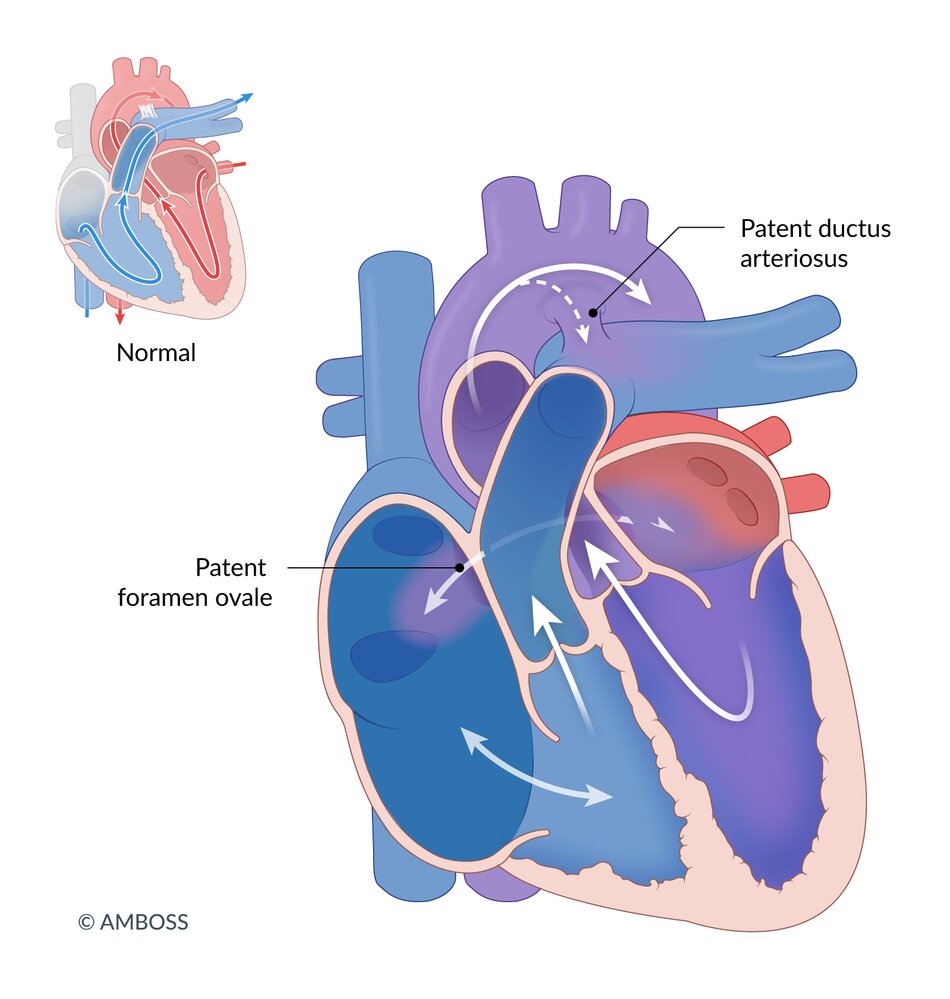
In the Ebstein anomaly, the tricuspid valve is displaced into the lumen of the right ventricle, resulting in atrialization of the right ventricle. The remaining ventricle is small and hypocontractile. Because tricuspid valve regurgitation is also usually present, there is retrograde flow in the right atrium, which is dilated by the volume overload, in addition to antegrade pulmonary blood flow. Blood flows through the patent foramen ovale or (commonly associated) atrial septal defect from the right atrium to the left atrium (right-to-left shunt) and then via the left ventricle into the circulation, resulting in pronounced cyanosis. The pulmonary circulation system may also be perfused through blood from the aorta (via the ductus arteriosus).
© AMBOSS
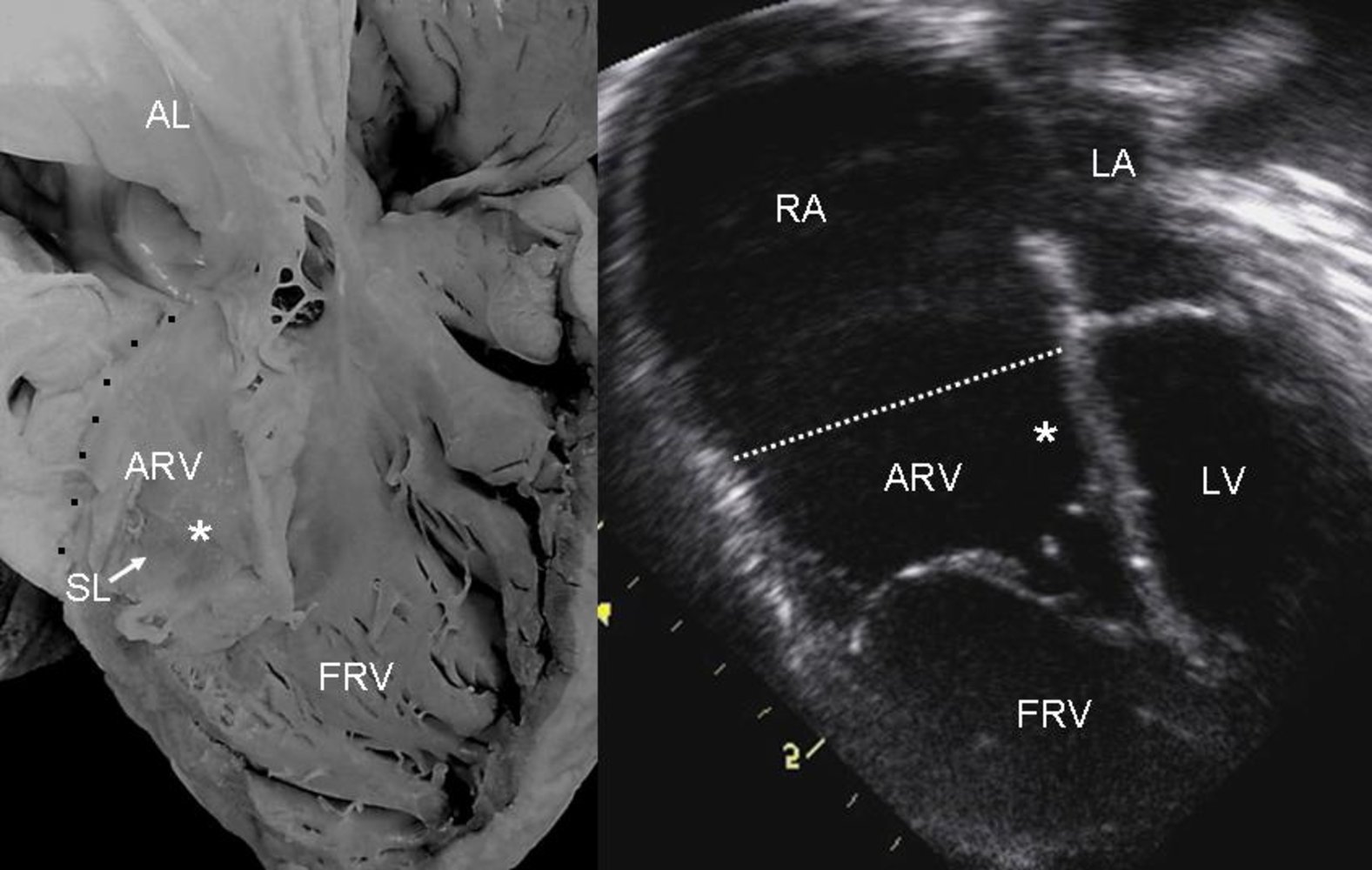
Photograph of an autopsied heart (left); ultrasound image of the heart (right)
- AL: elongated anterior leaflet of the tricuspid valve
- ARV: right ventricle
- FRV: functional right ventricle
- RA: right atrium
- SL: septal leaflet of the tricuspid valve
- *: tethering of the septal leaflet of the tricuspid valve to the ventricular wall
- LA: left atrium
- LV: left ventricle
- Dotted lines (black in the autopsied heart; white in the ultrasonographic image): identify the division between the RA and the RV
The leaflets of the tricuspid valve are tethered to the ventricular wall and displaced inferiorly into the lumen of the right ventricle. This results in the atrialization of the ARV, a small FRV, and a large RA.
Source: “Figure 4. in: Ebstein's Anomaly: Anatomo-echocardiographic correlation” by Luis Muñoz-Castellanos, Nilda Espinola-Zavaleta, Magdalena Kuri-Nivón & Candace Keirns, Cardiovascular Ultrasound Journal, BioMed Central, licensed under CC BY 2.0.
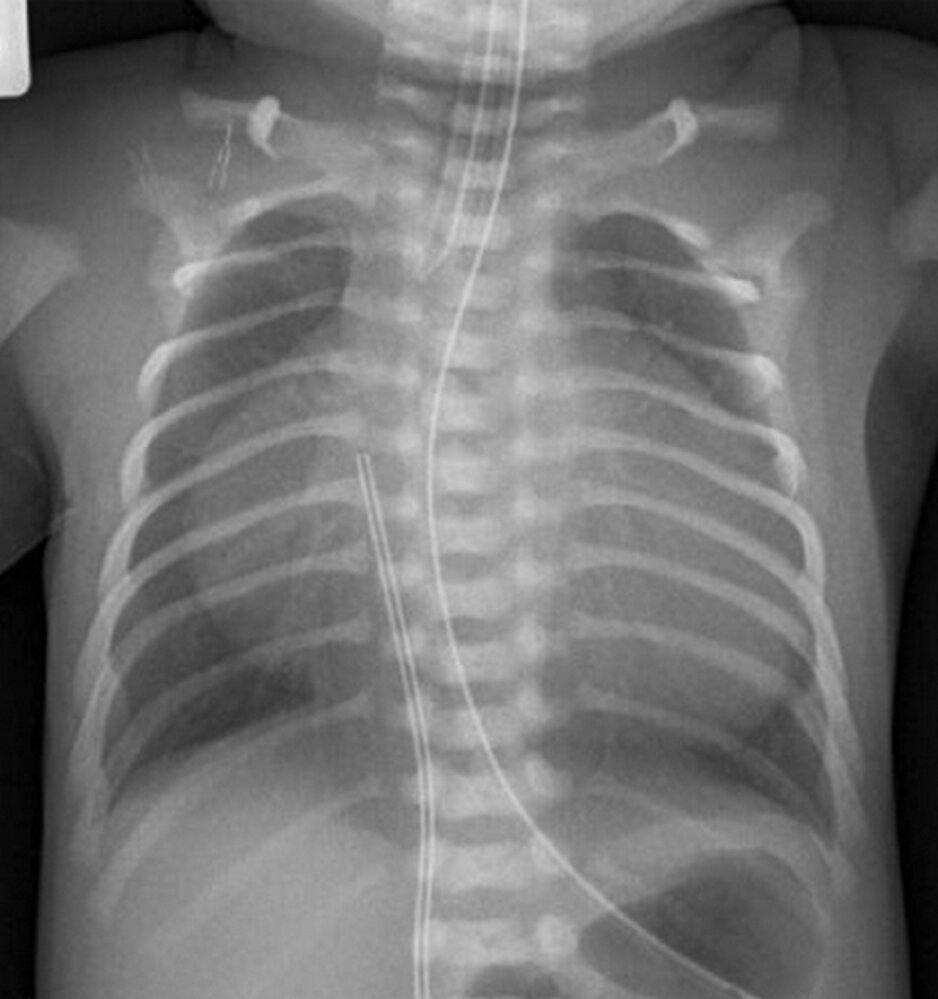
X-ray chest (PA view) of a neonate with Ebstein anomaly
The cardiac silhouette is markedly enlarged. Prominent convexity of the right heart border (green line) is the result of caudal displacement of the tricuspid valve with atrialization of the right ventricle and tricuspid regurgitation. The central pulmonary arteries (green arrowheads) and aortic knob (red arrowhead) are diminutive in appearance. An endotracheal tube is present, along with an umbilical venous catheter and nasogastric tube.
Radiographic findings in Ebstein anomaly may vary with the severity of the condition.
Source: “Figure 1, in: Pure Duplication of the Distal Long Arm of Chromosome 15 with Ebstein Anomaly and Clavicular Anomaly” by R. O'Connor A. Al-Murrani, S. Aftimos et al., Hindawi - Case Reports in Genetics, licensed under CC BY 3.0. The supplementary image with overlays of relevant areas was adapted from the image mentioned above (© AMBOSS).
Total anomalous pulmonary venous return (TAPVR)
Overview
- Definition: All four pulmonary veins drain into the systemic venous circulation (e.g., SVC, sinus venosus, RA, IVC) instead of the left atrium.
-
Other cardiac defects associated with TAPVR: allow for right-to-left shunting and the mixing of deoxygenated and oxygenated blood
- ASD
- PDA

Epidemiology
- Prevalence: 0.6–1.2/10,000 live births in the US [16]
Etiology
- Multifactorial (may be associated with genetic alterations) [69]
- Associated with heterotaxy syndromes that involve asplenia [70][71]
Pathophysiology
The severity of symptoms and clinical presentation is determined by the following factors:
- Venous mixing: oxygenated pulmonary venous return mixes with the systemic venous system → right-to-left shunting of partially oxygenated blood via an interatrial connection (e.g., ASD, PFO, rarely PDA) into the systemic arterial circulation → cyanosis
- Pulmonary venous obstruction: obstruction of pulmonary venous return → ↑ pulmonary venous pressure → ↑ pulmonary edema → ↑ PVR → RVH, ventricular dilation, and heart failure
Clinical features [72]
- Cyanosis
- Respiratory failure
- Poor feeding and failure to thrive
- Hepatomegaly
-
Cardiac examination findings
- Fixed split S2; or loud S2
- Systolic ejection murmur with diastolic rumble or mild to no murmur
Diagnostics
- Prenatal diagnosis: fetal echocardiography to detect abnormal pulmonary venous return [73]
-
Imaging
-
Echocardiography (confirmatory test)
- Malposition of pulmonary veins
- Enlarged right heart
- Right-to-left interatrial shunting
-
Chest x-ray
- “Snowman sign” (seen in supracardiac TAPVR): the orientation of the heart and superior mediastinum appear as the shape of a snowman
- Pulmonary hypervascularity
- Right heart enlargement
-
Echocardiography (confirmatory test)
-
ECG [72]
- Nonspecific findings
- Right axis deviation due to RVH
- Pulse oximetry: ↓ SpO2
Treatment
- Initial postnatal management: stabilization of cardiorespiratory status until surgery is performed (e.g., mechanical ventilation, supplemental O2, inotropes)
- Surgical repair: recommended in all patients regardless of the severity of disease [74]
Prognosis
- With surgical correction, the long-term survival rate is 80–90%. [75]
- Without treatment, ∼ 80% of patients die within the first year of life. [76]
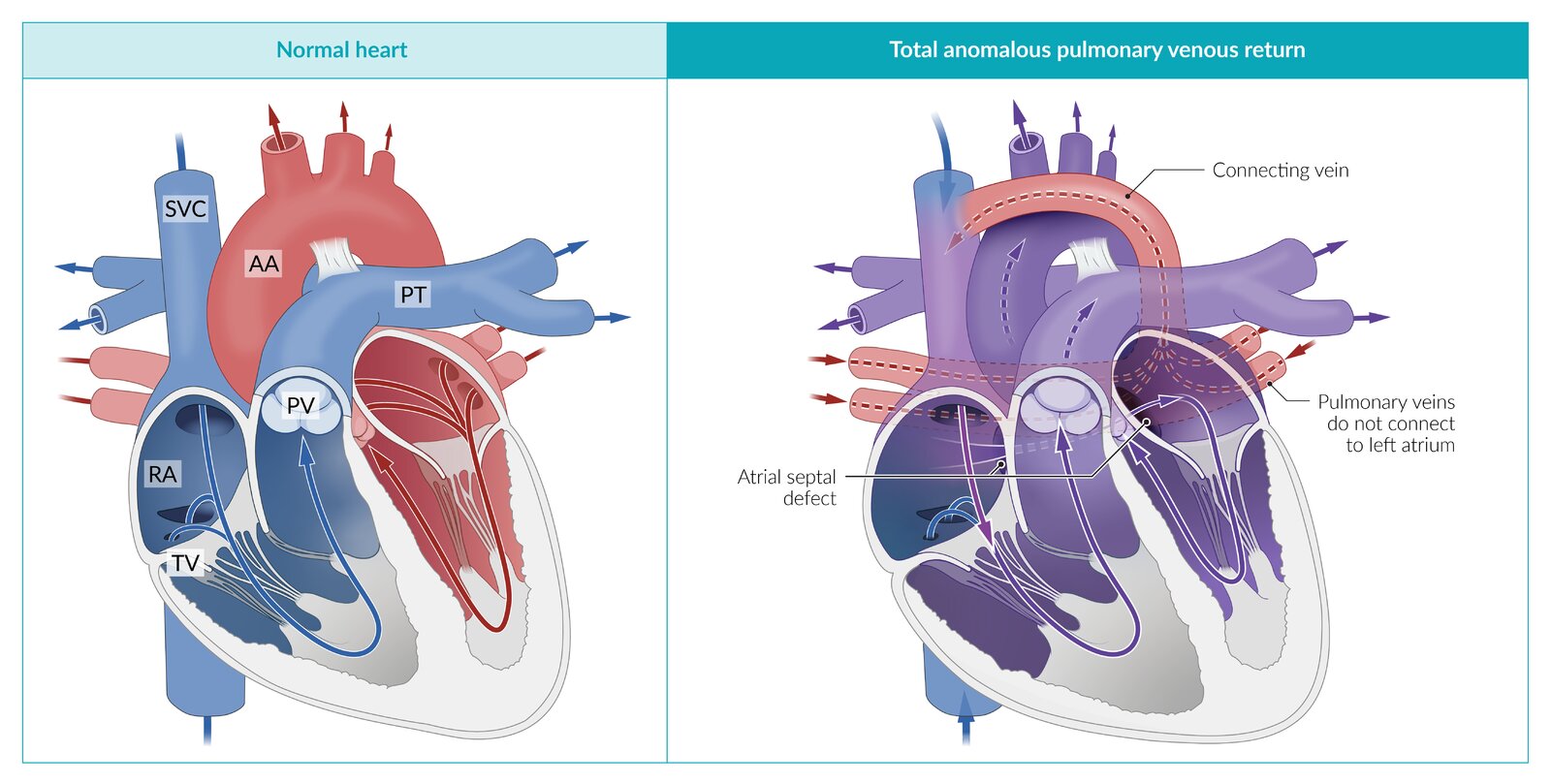
A congenital heart defect in which the pulmonary veins merge to form a connecting vein that drains into the systemic venous circulation (e.g., the vena cava) rather than the left atrium. Without return of blood to the left heart, there is no systemic circulation. Survival after birth depends on the presence of a right-to-left shunt (usually an atrial septal defect) to allow for blood to enter the left heart, then systemic circulation.
© AMBOSS
Persistent truncus arteriosus
Overview
- Definition: underdevelopment of the aorticopulmonary septum → failure of the truncus arteriosus to divide into the aorta and pulmonary trunk → a single trunk that receives output from both ventricles
-
Other cardiac defects associated with persistent truncus arteriosus
- VSD
- Interrupted aortic arch [77]
- Coronary artery anomalies [78]
Epidemiology
- Prevalence: ∼ 1/10,000 live births in the US [16]
Etiology
- The failure of neural crest cells to migrate during the development of the cardiac outflow tract results in incomplete AP septum formation. [79]
- Associated with DiGeorge syndrome [80][81]
Pathophysiology
- Deoxygenated and oxygenated blood mix via the VSD → the truncus arteriosus receiving both RV and LV output → blood flow to both pulmonary and systemic circulations
- The severity of disease is determined by pulmonary vascular resistance (PVR).
Clinical features
- Cyanosis
- Respiratory distress
- Failure to thrive
-
Cardiac examination findings
- Bounding peripheral pulses
- Harsh systolic murmur at the lower left sternal border
- Loud S2
Diagnostics
- Prenatal diagnosis: fetal echocardiography [82]
-
Imaging
- Echocardiography (confirmatory test): single overriding great vessel arising from the heart
-
Chest x-ray
- Right-sided aortic arch
- ↑ Pulmonary vascular markings
- Absent thymus, if associated with DiGeorge syndrome
- ECG: often normal and nonspecific findings
- Pulse oximetry: ↓ SpO2
Treatment
- Initial medical management: stabilization of cardiopulmonary function (e.g., diuretics, dopamine, ventilation, correction of metabolic acidosis) [9]
- Surgical repair: Surgical correction is recommended in the neonatal period. [83][84]
Prognosis
- With surgical repair, the 30-year survival rate is ∼ 70%. [85]

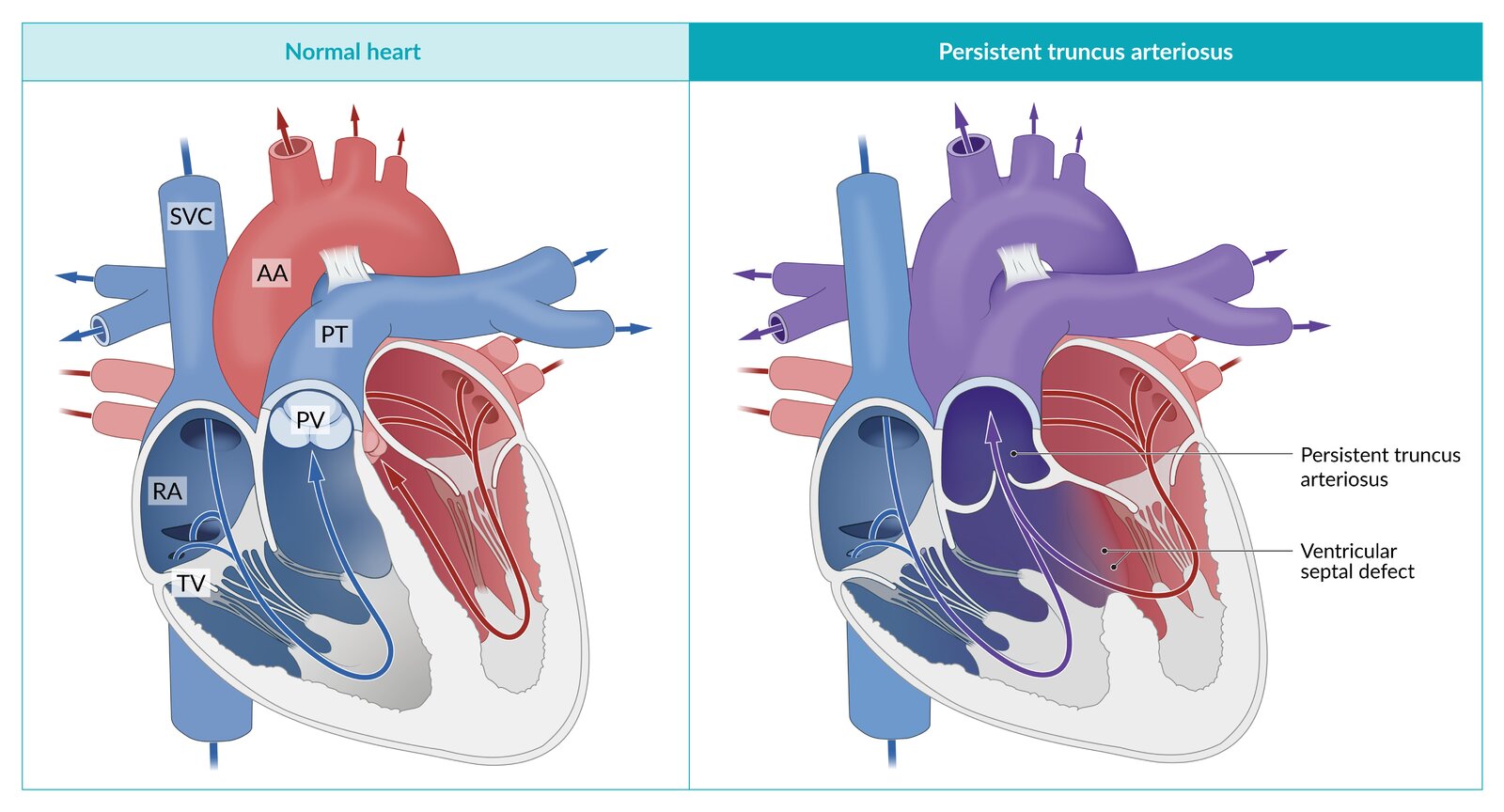
Persistent truncus arteriosus (PTA) is a congenital cyanotic heart defect in which the truncus arteriosus fails to divide into the pulmonary trunk and aorta and instead remains as a single trunk that receives blood from both ventricles. The pulmonary circulation and systemic circulation are not separated from one another, therefore, deoxygenated and oxygenated blood mix.
After birth, the pulmonary vascular resistance (PVR) drops, resulting in blood being shunted to the pulmonary circulation, leading to pulmonary congestion and subsequent heart failure.
© AMBOSS
Hypoplastic left heart syndrome (HLHS)
Overview
- Definition: spectrum of disease consisting of severe hypoplasia of the left ventricle with possible stenosis and/or atresia of the mitral valve, aortic valve, or aortic arch [86]
Epidemiology
- Extremely rare (2–3/10,000 live births in the US [16][87]
- Sex: ♂ > ♀ (1.5:1) [88]
- Although rare, HLHS is responsible for 25–40% of all neonatal cardiac deaths. [89]
Etiology
- Multifactorial: associated with single gene polymorphisms, altered blood flow patterns, and intrauterine infection [90][91][92]
- Syndromic associations: trisomy 13, trisomy 18, Jacobsen syndrome, and Turner syndrome [93][94]
Pathophysiology
- The hypoplastic left ventricle is nonfunctional, resulting in the RV becoming the primary supply for pulmonary and systemic circulations.
- Survival depends on the presence of a PDA (right-to-left shunt); and an ASD.

Clinical findings
- May be asymptomatic at birth if the open PDA provides adequate systemic perfusion
- Aggravation of symptoms following closure of the PDA
- Postnatal cyanosis (not corrected by supplement O2)
- Tachypnea
- ↓ BP, ↓ peripheral pulses, and cool extremities
- Heart failure and cardiogenic shock
-
Cardiac examination findings
- Typically no murmur
- Loud, single S2
Diagnostics
- Prenatal diagnosis: fetal echocardiography [95]
-
Imaging
- Echocardiography (confirmatory test): visualization of absent or hypoplastic LV
- Chest x-ray: variable and nonspecific findings
- ECG: typically normal
- Pulse oximetry: ↓ SpO2
Treatment
- Initial medical management: continuous PGE1 infusion prior to heart surgery [96]
-
Surgical repair: Palliative surgery is used in single-ventricular cardiopathologies. [88]
-
Three-step staged surgical correction
- Norwood procedure (stage I): performed while the patient is a neonate [97][98]
- Glenn procedure (stage II): performed at ∼ 3–6 months of age [99]
- Fontan procedure (stage III): performed after 2–3 years of age [100]
- Alternative: heart transplant [101]
-
Three-step staged surgical correction
Prognosis
- Even with surgical correction, the 5-year survival rate remains low (∼ 65%). [97]
- For patients that survive to one year of age, long-term survival (up to 18 years of age) is ∼ 90%. [102][103]
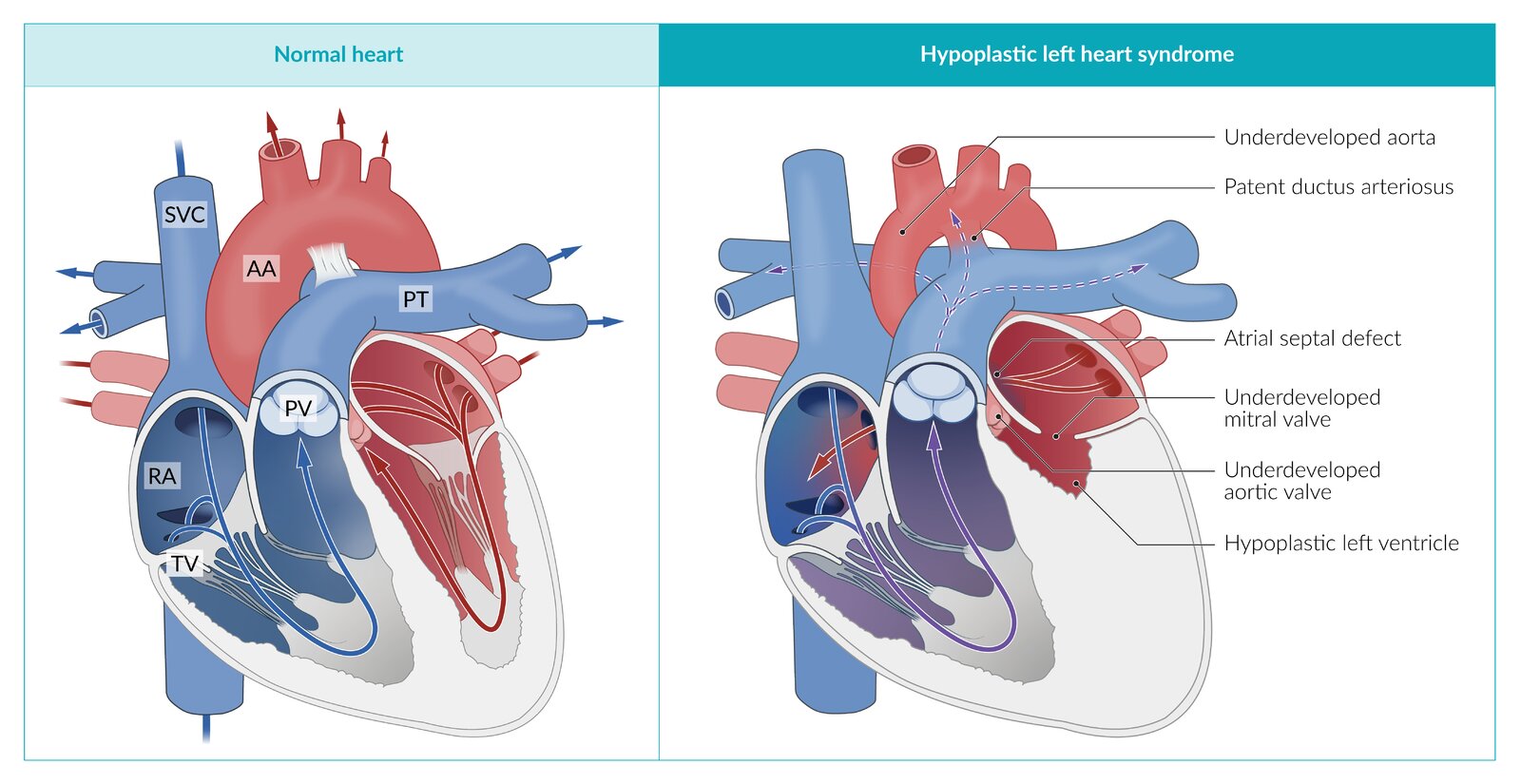
Hypoplastic left heart syndrome is a cyanotic congenital heart defect in which the left ventricle (LV) does not form properly, resulting in an absent or hypoplastic LV. Stenosis and/or atresia of the mitral valve, aortic valve and/or aortic arch are also possible.
The left ventricle is nonfunctional, resulting in the RV becoming the primary supply for the pulmonary and systemic circulations. An atrial septum defect ensures that oxygenated blood from the lungs flows from the left atrium into the right atrium. A right-to-left shunt across the patent ductus arteriosus then ensures that blood flows from the main pulmonary artery to the aorta, to reach the systemic circulation.
© AMBOSS
References
- de-Wahl Granelli A, Wennergren M, Sandberg K, et al. "Impact of pulse oximetry screening on the detection of duct dependent congenital heart disease: a Swedish prospective screening study in 39 821 newborns". BMJ. 338(jan08 2). :a3037-a3037. (2009)
- Mahle WT, Newburger JW, Matherne GP, et al. "Role of Pulse Oximetry in Examining Newborns for Congenital Heart Disease: A Scientific Statement from the AHA and AAP". Pediatrics. 124(2). :823-836. (2009)
- Wren C, Richmond S, Donaldson L. "Presentation of congenital heart disease in infancy: implications for routine examination". Archives of Disease in Childhood - Fetal and Neonatal Edition. 80(1). :F49-F53. (1999)
- Liberman RF, Getz KD, Lin AE, et al. "Delayed Diagnosis of Critical Congenital Heart Defects: Trends and Associated Factors". Pediatrics. 134(2). :e373-e381. (2014)
- Donofrio MT, Moon-Grady AJ, Hornberger LK, et al. "Diagnosis and Treatment of Fetal Cardiac Disease". Circulation. 129(21). :2183-2242. (2014)
- Olley PM, Coceani F, Bodach E. "E-type prostaglandins: a new emergency therapy for certain cyanotic congenital heart malformations.". Circulation. 53(4). :728-731. (1976)
- Walls R, Hockberger R, Gausche-Hill M, Erickson TB, Wilcox SR. "Rosen's Emergency Medicine 10th edition- Concepts and Clinical Practice E-Book". Elsevier Health Sciences. (2022). ISBN: 9780323757904
- Akkinapally S, Hundalani SG, Kulkarni M, et al. "Prostaglandin E1 for maintaining ductal patency in neonates with ductal-dependent cardiac lesions". Cochrane Database Syst Rev. (2018)
- Kirk R, Dipchand AI, Rosenthal DN, et al. "The International Society for Heart and Lung Transplantation Guidelines for the management of pediatric heart failure: Executive summary". J Heart Lung Transplant. 33(9). :888-909. (2014)
- Wilson W, Taubert KA, Gewitz M, et al. "Prevention of Infective Endocarditis Guidelines From the American Heart Association". Circulation. (2007)
- Rushani D, Kaufman JS, Ionescu-Ittu R, et al. "Infective Endocarditis in Children With Congenital Heart Disease". Circulation. 128(13). :1412-1419. (2013)
- O’Brien P, Marshall AC. "Tetralogy of Fallot". Circulation. 130(4). (2014)
- Braun-Falco M, Mankin HJ, Wenger SL, et al. "Pentalogy of Fallot". Springer Berlin Heidelberg. :1602-1605. (2009). ISBN: 9783540671367
- Dabizzi RP, Caprioli G, Aiazzi L, et al. "Distribution and anomalies of coronary arteries in tetralogy of fallot.". Circulation. 61(1). :95-102. (1980)
- Centers for Disease Control and Prevention (CDC).. "Improved national prevalence estimates for 18 selected major birth defects--United States, 1999-2001.". MMWR Morb Mortal Wkly Rep. 54(51). :1301-5. (2006)
- Reller MD, Strickland MJ, Riehle-Colarusso T, Mahle WT, Correa A. "Prevalence of Congenital Heart Defects in Metropolitan Atlanta, 1998-2005". J Pediatr. 153(6). :807-813. (2008)
- Greenway SC, Pereira AC, Lin JC, et al. "De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot". Nat Genet. 41(8). :931-935. (2009)
- Goldmuntz E, Clark BJ, Mitchell LE, et al. "Frequency of 22q11 deletions in patients with conotruncal defects". J Am Coll Cardiol. 32(2). :492-498. (1998)
- Kessler-Icekson G, Birk E, Weintraub AY, et al. "Association of tetralogy of Fallot with a distinct region of del22q11.2". Am J Med Genet. 107(4). :294-298. (2002)
- Freeman SB, Taft LF, Dooley KJ, et al. "Population-based study of congenital heart defects in Down syndrome.". Am J Med Genet. 80(3). :213-7. (1998)
- Claire A Irving, Milind P Chaudhari. "Cardiovascular abnormalities in Down's syndrome: spectrum, management and survival over 22 years". Arch Dis Child. 97(4). :326-330. (2011)
- Lenke RR, Levy HL. "Maternal Phenylketonuria and Hyperphenylalaninemia". N Engl J Med. 303(21). :1202-1208. (1980)
- Platt LD, Koch R, Hanley WB, et al. "The International Study of Pregnancy Outcome in Women with Maternal Phenylketonuria: Report of a 12-year study". Am J Obstet Gynecol. 182(2). :326-333. (2000)
- Lisowski LA, Verheijen PM, Copel JA, et al. "Congenital Heart Disease in Pregnancies Complicated by Maternal Diabetes Mellitus". Herz. 35(1). :19-26. (2010)
- Ray JG. "Preconception care and the risk of congenital anomalies in the offspring of women with diabetes mellitus: a meta-analysis". QJM. 94(8). :435-444. (2001)
- "Tetralogy of Fallot". http://www.msdmanuals.com/professional/pediatrics/congenital-cardiovascular-anomalies/tetralogy-of-fallot. [2018-11-01]
- "Tetralogy of Fallot". https://www.mayoclinic.org/diseases-conditions/tetralogy-of-fallot/symptoms-causes/syc-20353477#:~:text=Sometimes%2C%20babies%20who%20have%20tetralogy,2%20to%204%20months%20old.. [2020-01-01]
- Kothari SS. "Mechanism of cyanotic spells in tetralogy of Fallot — the missing link?". Int J Cardiol. 37(1). :1-5. (1992)
- Duro RP, Moura C, Leite-Moreira A. "Anatomophysiologic basis of tetralogy of Fallot and its clinical implications.". Rev Port Cardiol. 29(4). :591-630. (2010)
- Bhat AH, Kehl DW, Tacy TA, Moon-Grady AJ, Hornberger LK. "Diagnosis of Tetralogy of Fallot and Its Variants in the Late First and Early Second Trimester: Details of Initial Assessment and Comparison with Later Fetal Diagnosis". Echocardiography. 30(1). :81-87. (2012)
- Stout KK, Daniels CJ, Aboulhosn JA, et al. "2018 AHA/ACC Guideline for the Management of Adults With Congenital Heart Disease". J Am Coll Cardiol. 73(12). :e81-e192. (2019)
- Moller JH, Johnson WH. "Pediatric Cardiology". Wiley-Blackwell. (2008). ISBN: 9781405178181
- Al Habib HF, Jacobs JP, Mavroudis C, et al. "Contemporary Patterns of Management of Tetralogy of Fallot: Data From The Society of Thoracic Surgeons Database". Ann Thorac Surg. 90(3). :813-820. (2010)
- Sarris GE, Comas JV, Tobota Z, Maruszewski B. "Results of reparative surgery for tetralogy of Fallot: data from the European Association for Cardio-Thoracic Surgery Congenital Database". Eur J Cardiothorac Surg. 42(5). :766-774. (2012)
- Blalock A. "Landmark article May 19, 1945: The surgical treatment of malformations of the heart in which there is pulmonary stenosis or pulmonary atresia. By Alfred Blalock and Helen B. Taussig". JAMA. 251(16). :2123-2138. (1984)
- Kiran U, Aggarwal S, Choudhary A, Uma B, Kapoor PM. "The blalock and taussig shunt revisited.". Ann Card Anaesth. 20(3). :323-330
- Bertranou EG, Blackstone EH, Hazelrig JB, Turner ME, Kirklin JW. "Life expectancy without surgery in tetralogy of fallot". Am J Cardiol. 42(3). :458-466. (1978)
- Park CS, Lee JR, Lim H-G, Kim W-H, Kim YJ. "The long-term result of total repair for tetralogy of Fallot☆". Eur J Cardiothorac Surg. 38(3). :311-317. (2010)
- Warnes CA. "Transposition of the Great Arteries". Circulation. 114(24). :2699-2709. (2006)
- Corno AF. "Complete transposition of the great arteries". Steinkopff. :115-135. (2003). ISBN: 9783642632457
- Momma K. "Cardiovascular Anomalies Associated With Chromosome 22q11.2 Deletion Syndrome". Am J Cardiol. 105(11). :1617-1624. (2010)
- Squarcia U, Macchi C. "Transposition of the great arteries". Curr Opin Pediatr. 23(5). :518-522. (2011)
- Ravi P, Mills L, Fruitman D, et al. "Population trends in prenatal detection of transposition of great arteries: impact of obstetric screening ultrasound guidelines". Ultrasound Obstet Gynecol. 51(5). :659-664. (2018)
- Sarris GE, Balmer C, Bonou P, et al. "Clinical guidelines for the management of patients with transposition of the great arteries with intact ventricular septum". Eur J Cardiothorac Surg. 51(1). :e1-e32. (2017)
- Freed MD, Heymann MA, Lewis AB, Roehl SL, Kensey RC. "Prostaglandin E1 infants with ductus arteriosus-dependent congenital heart disease.". Circulation. 64(5). :899-905. (1981)
- Hiremath G, Natarajan G, Math D, Aggarwal S. "Impact of balloon atrial septostomy in neonates with transposition of great arteries". J Perinatol. 31(7). :494-499. (2011)
- Van der Laan ME, Verhagen EA, Bos AF, Berger RMF, Kooi EMW. "Effect of balloon atrial septostomy on cerebral oxygenation in neonates with transposition of the great arteries". Pediatr Res. 73(1). :62-67. (2012)
- Feltes TF, Bacha E, Beekman RH, et al. "Indications for Cardiac Catheterization and Intervention in Pediatric Cardiac Disease". Circulation. 123(22). :2607-2652. (2011)
- Anderson BR, Ciarleglio AJ, Hayes DA, et al. "Earlier Arterial Switch Operation Improves Outcomes and Reduces Costs for Neonates With Transposition of the Great Arteries". J Am Coll Cardiol. 63(5). :481-487. (2014)
- Lim JM, Porayette P, Marini D, et al. "Associations Between Age at Arterial Switch Operation, Brain Growth, and Development in Infants With Transposition of the Great Arteries". Circulation. 139(24). :2728-2738. (2019)
- P.A. Hutter, D.L. Kreb, S.F. Mantel, et al. "Twenty-five years' experience with the arterial switch operation". J Thorac Cardiovasc Surg. 124(4). :790-797. (2002)
- Jatene AD, Fontes VF, Paulista PP, et al. "Anatomic correction of transposition of the great vessels.". J Thorac Cardiovasc Surg. 72(3). :364-70. (1976)
- RASTELLI GC, WALLACE RB, ONGLEY PA. "Complete Repair of Transposition of the Great Arteries with Pulmonary Stenosis". Circulation. 39(1). :83-95. (1969)
- Navabi MA, Shabanian R, Kiani A, Rahimzadeh M. "The effect of ventricular septal defect enlargement on the outcome of Rastelli or Rastelli-type repair". J Thorac Cardiovasc Surg. 138(2). :390-396. (2009)
- Driscoll DJ, Shaddy RE, Feltes TF. "Moss & Adams Heart Disease in Infants, Children, and Adolescents". Lippincott Williams & Wilkins. (2012). ISBN: 9781451118933
- Tchervenkov CI, Jacobs ML, Tahta SA. "Congenital Heart Surgery Nomenclature and Database Project: hypoplastic left heart syndrome". Ann Thorac Surg. 69(3). :170-179. (2000)
- Gordon BM, Rodriguez S, Lee M, Chang R-K. "Decreasing Number of Deaths of Infants with Hypoplastic Left Heart Syndrome". J Pediatr. 153(3). :354-358. (2008)
- Karamlou T, Diggs BS, Ungerleider RM, Welke KF. "Evolution of treatment options and outcomes for hypoplastic left heart syndrome over an 18-year period". J Thorac Cardiovasc Surg. 139(1). :119-127. (2010)
- Yabrodi M, Mastropietro CW. "Hypoplastic left heart syndrome: from comfort care to long-term survival.". Pediatr Res. 81(1-2). :142-149. (2017)
- Mäkikallio K, McElhinney DB, Levine JC, et al. "Fetal Aortic Valve Stenosis and the Evolution of Hypoplastic Left Heart Syndrome". Circulation. 113(11). :1401-1405. (2006)
- McElhinney DB, Geiger E, Blinder J, Woodrow Benson D, Goldmuntz E. "NKX2.5mutations in patients with congenital heart disease". J Am Coll Cardiol. 42(9). :1650-1655. (2003)
- Hinton RB, Martin LJ, Rame-Gowda S, et al. "Hypoplastic Left Heart Syndrome Links to Chromosomes 10q and 6q and Is Genetically Related to Bicuspid Aortic Valve". J Am Coll Cardiol. 53(12). :1065-1071. (2009)
- Benson DW, Martin LJ, Lo CW. "Genetics of Hypoplastic Left Heart Syndrome". J Pediatr. 173. :25-31. (2016)
- Mattina T, Perrotta CS, Grossfeld P. "Jacobsen syndrome". Orphanet J Rare Dis. 4(1). (2009)
- Brown DW, Cohen KE, O’Brien P, et al. "Impact of Prenatal Diagnosis in Survivors of Initial Palliation of Single Ventricle Heart Disease". Pediatr Cardiol. 36(2). :314-321. (2014)
- Hoque T, Richmond M, Vincent JA, Bacha E, Torres A. "Current Outcomes of Hypoplastic Left Heart Syndrome With Restrictive Atrial Septum: A Single-Center Experience". Pediatr Cardiol. 34(5). :1181-1189. (2013)
- Newburger JW, Sleeper LA, Gaynor JW, et al. "Transplant-Free Survival and Interventions at 6 Years in the SVR Trial". Circulation. 137(21). :2246-2253. (2018)
- Ohye RG, Sleeper LA, Mahony L, et al. "Comparison of Shunt Types in the Norwood Procedure for Single-Ventricle Lesions". N Engl J Med. 362(21). :1980-1992. (2010)
- Meza JM, Hickey EJ, Blackstone EH, et al. "The Optimal Timing of Stage 2 Palliation for Hypoplastic Left Heart Syndrome". Circulation. 136(18). :1737-1748. (2017)
- Hirsch JC, Goldberg C, Bove EL, et al. "Fontan Operation in the Current Era". Ann Surg. 126. :52-60. (2008)
- Alsoufi B, Mahle WT, Manlhiot C, et al. "Outcomes of heart transplantation in children with hypoplastic left heart syndrome previously palliated with the Norwood procedure". J Thorac Cardiovasc Surg. 151(1). :167-175.e2. (2016)
- Alsoufi B, Mori M, Gillespie S, et al. "Impact of Patient Characteristics and Anatomy on Results of Norwood Operation for Hypoplastic Left Heart Syndrome". Ann Thorac Surg. 100(2). :591-598. (2015)
- Siffel C, Riehle-Colarusso T, Oster ME, Correa A. "Survival of Children With Hypoplastic Left Heart Syndrome". Pediatrics. 136(4). :e864-e870. (2015)
- Sittiwangkul R, Azakie A, Van Arsdell GS, Williams WG, McCrindle BW. "Outcomes of tricuspid atresia in the Fontan era". Ann Thorac Surg. 77(3). :889-894. (2004)
- Wald RM, Tham EB, McCrindle BW, et al. "Outcome after prenatal diagnosis of tricuspid atresia: A multicenter experience". Am Heart J. 153(5). :772-778. (2007)
- Rao PS. "Tricuspid atresia". Curr Treat Options Cardiovasc Med. 2(6). :507-520. (2000)
- Bleyl SB, Saijoh Y, Bax NAM, et al. "Dysregulation of the PDGFRA gene causes inflow tract anomalies including TAPVR: integrating evidence from human genetics and model organisms". Hum Mol Genet. 19(7). :1286-1301. (2010)
- Bartz PJ, Driscoll DJ, Dearani JA, et al. "Early and Late Results of the Modified Fontan Operation for Heterotaxy Syndrome". J Am Coll Cardiol. 48(11). :2301-2305. (2006)
- Virmani R, Carter-Monroe N, Taylor AJ. "Congenital Anomalies and Malformations of the Vasculature". Elsevier. :771-789. (2013). ISBN: 9781437729306
- GATHMAN GE, NADAS AS. "Total Anomalous Pulmonary Venous Connection". Circulation. 42(1). :143-154. (1970)
- Ganesan S, Brook MM, Silverman NH, Moon-Grady AJ. "Prenatal Findings in Total Anomalous Pulmonary Venous Return". J Ultrasound Med. 33(7). :1193-1207. (2014)
- Hancock Friesen CL, Zurakowski D, Thiagarajan RR, et al. "Total Anomalous Pulmonary Venous Connection: An Analysis of Current Management Strategies in a Single Institution". Ann Thorac Surg. 79(2). :596-606. (2005)
- Bando K, Turrentine MW, Ensing GJ, et al. "Surgical management of total anomalous pulmonary venous connection. Thirty-year trends.". Circulation. 94(9 Suppl). :II12-6. (1996)
- Burroughs JT, Edwards JE. "Total anomalous pulmonary venous connection". Am Heart J. 59(6). :913-931. (1960)
- Konstantinov IE, Karamlou T, Blackstone EH, et al. "Truncus Arteriosus Associated with Interrupted Aortic Arch in 50 Neonates: A Congenital Heart Surgeons Society Study". Ann Thorac Surg. 81(1). :214-222. (2006)
- De la Cruz MV, Cayre R, Angelini P, Noriega-Ramos N, Sadowinski S. "Coronary arteries in truncus arteriosus". Am J Cardiol. 66(20). :1482-1486. (1990)
- Scholl AM, Kirby ML. "Signals controlling neural crest contributions to the heart". Wiley Interdiscip Rev Syst Biol Med. 1(2). :220-227. (2009)
- Ziolkowska L, Kawalec W, Turska-Kmiec A, et al. "Chromosome 22q11.2 microdeletion in children with conotruncal heart defects: frequency, associated cardiovascular anomalies, and outcome following cardiac surgery". Eur J Pediatr. 167(10). :1135-1140. (2008)
- Botto LD, May K, Fernhoff PM, et al. "A Population-Based Study of the 22q11.2 Deletion: Phenotype, Incidence, and Contribution to Major Birth Defects in the Population". Pediatrics. 112(1). :101-107. (2003)
- Swanson TM, Selamet Tierney ES, Tworetzky W, Pigula F, McElhinney DB. "Truncus Arteriosus: Diagnostic Accuracy, Outcomes, and Impact of Prenatal Diagnosis". Pediatr Cardiol. 30(3). :256-261. (2008)
- Rodefeld MD, Hanley FL. "Neonatal truncus arteriosus repair: Surgical techniques and clinical management". Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 5(1). :212-217. (2002)
- Thompson LD, McElhinney DB, Reddy VM, et al. "Neonatal repair of truncus arteriosus: continuing improvement in outcomes". Ann Thorac Surg. 72(2). :391-395. (2001)
- Naimo PS, Fricke TA, Yong MS, et al. "Outcomes of Truncus Arteriosus Repair in Children: 35 Years of Experience From a Single Institution". Semin Thorac Cardiovasc Surg. 28(2). :500-511. (2016)
- Attenhofer Jost CH, Connolly HM, Dearani JA, Edwards WD, Danielson GK. "Ebstein's Anomaly". Circulation. 115. :277-285. (2007)
- J. Hebe. "Ebstein's Anomaly in Adults.Arrhythmias: Diagnosis and Therapeutic Approach". Thorac Cardiovasc Surg. 48(4). :214-219. (2000)
- Lupo PJ, Langlois PH, Mitchell LE. "Epidemiology of Ebstein anomaly: Prevalence and patterns in Texas, 1999-2005". Am J Med Genet A. 155(5). :1007-1014. (2011)
- Patorno E, Huybrechts KF, Bateman BT, et al. "Lithium Use in Pregnancy and the Risk of Cardiac Malformations". N Engl J Med. 376(23). :2245-2254. (2017)
- Cohen LS. "A Reevaluation of Risk of In Utero Exposure to Lithium". JAMA. 271(2). :146. (1994)
- McKnight RF, Adida M, Budge K, et al. "Lithium toxicity profile: a systematic review and meta-analysis". Lancet. 379(9817). :721-728. (2012)
- David S. Celermajer, Catherine Bull, Janice A. Till, et al. "Ebstein's anomaly: Presentation and outcome from fetus to adult". J Am Coll Cardiol. 23(1). :170-176. (1994)
- Knott-Craig CJ, Goldberg SP, Overholt ED, Colvin EV, Kirklin JK. "Repair of Neonates and Young Infants With Ebstein’s Anomaly and Related Disorders". Ann Thorac Surg. 84(2). :587-593. (2007)
- Stout KK, Daniels CJ, Aboulhosn JA, et al. "2018 AHA/ACC Guideline for the Management of Adults With Congenital Heart Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines". Circulation. 139(14). (2019)
- JONATHAN D. REICH, DEBBIE AULD, EDWARD HULSE, KEVIN SULLIVAN, ROBERT CAMPBELL. "The Pediatric Radiofrequency Ablation Registry's Experience with Ebstein's Anomaly". J Cardiovasc Electrophysiol. 9(12). :1370-1377. (1998)
- Brunicardi F, Andersen D, Billiar T, et al. "Schwartz's Principles of Surgery". McGraw-Hill Education. (2014). ISBN: 9780071800921
- Altman CA. "Identifying newborns with critical congenital heart disease". UpToDate. UpToDate. https://www.uptodate.com/contents/identifying-newborns-with-critical-congenital-heart-disease. [2018-06-14]
- Geggel RL. "Cardiac causes of cyanosis in the newborn". UpToDate. UpToDate. https://www.uptodate.com/contents/cardiac-causes-of-cyanosis-in-the-newborn. [2018-05-13]