Summary
Cystic fibrosis (CF) is an autosomal recessive disorder that is common in individuals of European descent. It is caused by mutations in the CFTR gene, which encodes the CF transmembrane conductance regulator (CFTR) protein. These mutations result in defective chloride (Cl-) channels. Mandated newborn screening (NBS) in many countries can frequently detect CF during the asymptomatic newborn period. Initial manifestations may include meconium ileus, failure to thrive, and symptoms of malabsorption, and later in life, patients may experience symptoms such as recurrent respiratory infections, recurrent pancreatitis, and infertility. As the disease progresses, defective Cl- channels in the respiratory tract result in thickened bronchial mucus and impaired mucociliary clearance, which leads to chronic respiratory infections, pulmonary colonization with multiresistant bacteria, and a progressive decline in lung function. Patients often experience acute episodes of worsening respiratory symptoms, known as pulmonary exacerbations. CFTR dysfunction in the exocrine pancreas and biliary tract can ultimately lead to CF-related liver disease (CFLD) and CF-related diabetes (CFRD). If CF is suspected, the first diagnostic study is a sweat test, usually followed by genetic testing, and if the diagnosis remains unclear, CFTR physiologic testing. Patients with CF should be managed at a CF-accredited center, if possible. Management should focus on slowing the progression of lung disease, eradicating and/or suppressing chronic infections, preventing and/or treating exacerbations, and addressing complications and comorbidities. Many patients may also benefit from novel treatments such as CFTR modulators, which can partially restore CFTR function. With the recent advances in treatment, the predicted median life expectancy of a patient born in 2021 with CF is estimated to be ∼ 45 years. Complications due to lung disease are the most common cause of death in CF.

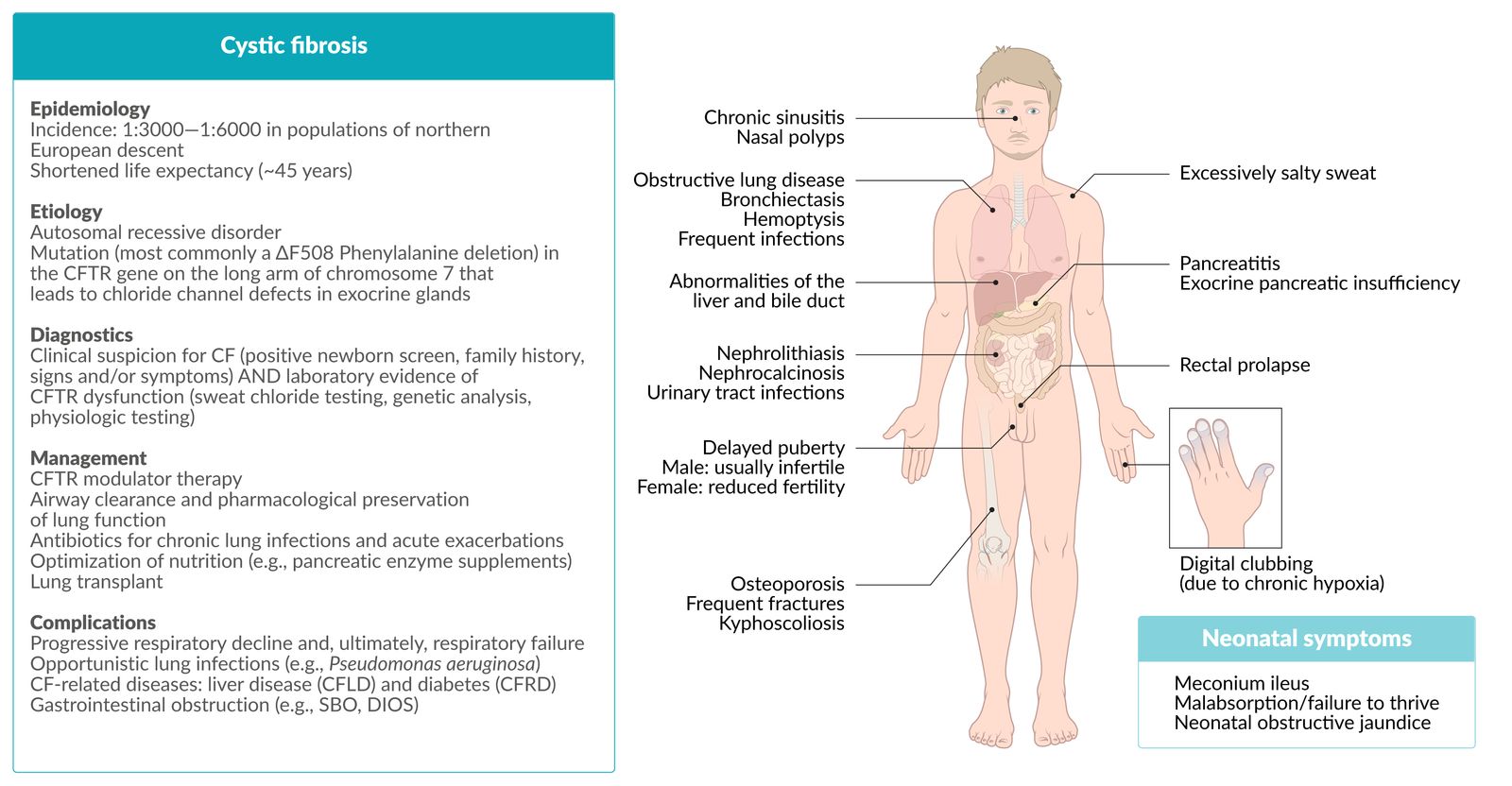
CF: cystic fibrosis; CFTR: CF transmembrane conductance regulator; CFLD: CF-related liver disease; CFRD: CF-related diabetes; SBO: small bowel obstruction; DIOS: distal intestinal obstruction syndrome
© AMBOSS
Epidemiology
- Second most common genetic metabolic disorder in individuals of Northern European descent after hemochromatosis [1][2][3]
- Incidence: approx. 1:3500 in the US [4]
Epidemiological data refers to the US, unless otherwise specified.
Etiology
- CF is a hereditary autosomal recessive disorder caused by defective CFTR (cystic fibrosis transmembrane conductance regulator) protein due to mutation in the CFTR gene located on the long arm of chromosome 7. [5]
- The most common mutation causing CF is delta F508 (ΔF508), a codon deletion that leads to the absence of phenylalanine (F) in position 508 of the CFTR protein.
Children whose parents are both heterozygous carriers of cystic fibrosis have a 25% chance of being affected by the condition.
Pathophysiology
-
General considerations
- The CFTR gene encodes the CFTR protein, which is an important component of the ATP-gated chloride channel in cell membranes.
- Mutated CFTR gene → misfolded protein → retention for degradation of the defective protein in the rough endoplasmic reticulum (rER) → absence of ATP-gated chloride channel on the cell surface of epithelial cells throughout the body (e.g., intestinal and respiratory epithelia, sweat glands, exocrine pancreas, exocrine glands of reproductive organs) [6][7]
-
In sweat glands
- The chloride channel is responsible for transporting Cl- from the lumen into the cell (reabsorption).
- Defective ATP-gated chloride channel → inability to reabsorb Cl- from the lumen of the sweat glands → reduced reabsorption of Na+ and H2O → excessive loss of salt and elevated levels of NaCl in sweat
-
In all other exocrine glands (e.g., in the GI tract or lungs)
- The chloride channel is responsible for transporting Cl- from the cell into the lumen (secretion).
- Defective ATP-gated chloride channel → inability to transport intracellular Cl- across the cell membrane → reduced secretion of Cl- and H2O → accumulation of intracellular Cl- → ↑ Na+ reabsorption (via ENaC); → ↑ H2O reabsorption; → formation of hyperviscous mucus → accumulation of secretions and blockage of small passages of affected organs → chronic inflammation and remodeling → organ damage (see “Clinical features” below)
- ↑ Na+ reabsorption → transepithelial potential difference between interstitial fluid and the epithelial surface increases; (i.e., negative charge increases; e.g., from normal -13 mv to abnormal -25 mv)

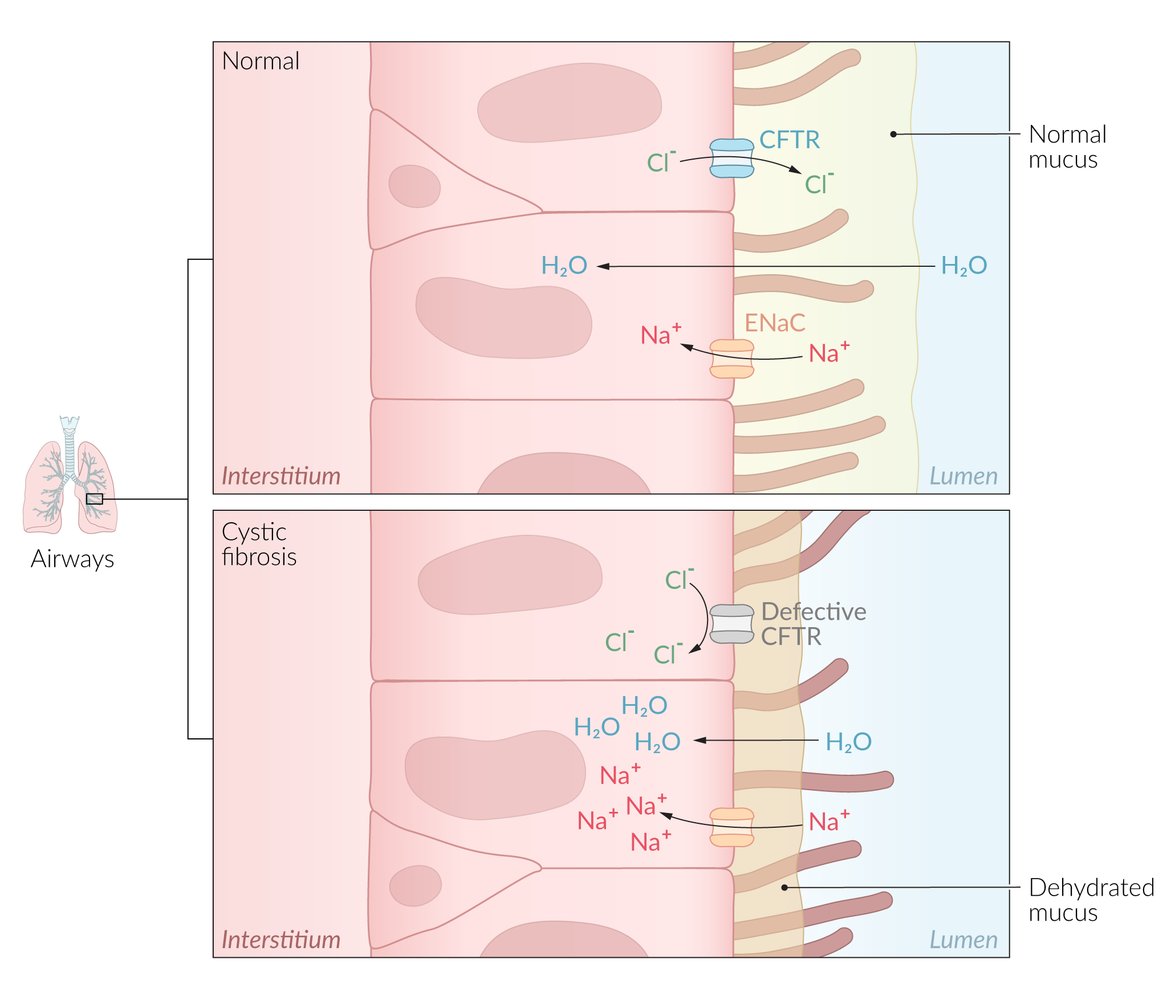
A mutation in the CFTR gene results in defective protein synthesis and the absence of the ATP-gated chloride channel in the cell surface of epithelial cells throughout the body (intestinal and respiratory epithelia, sweat glands, exocrine pancreas, exocrine glands, and reproductive organs). Chloride and sodium are normally in equilibrium, but the accumulation of intracellular chloride due to the absence of a transporter results in the retention of intracellular sodium and water (due to osmosis). This ultimately results in hyperviscous mucus, which causes blockages of small organs and airways, inflammation, and end-organ damage.
© AMBOSS
Definitions
- CF pulmonary exacerbations: episodes of acute worsening of respiratory symptoms (e.g., increased cough and sputum production, fever, dyspnea, hemoptysis) in patients with CF, which can result in an irreversible decline in lung function [8]
- CF-related diabetes (CFRD): Pancreatic exocrine dysfunction leads to pancreatic damage, which destroys the pancreatic endocrine insulin-producing beta cells and results in insulin deficiency. [9]
- CF-related liver disease (CFLD): Exocrine complication of CF that can include neonatal obstructive cholestasis, cholelithiasis, biliary cirrhosis, portal hypertension, and end-stage liver disease [10]
Clinical features

Gastrointestinal symptoms of CF [11]
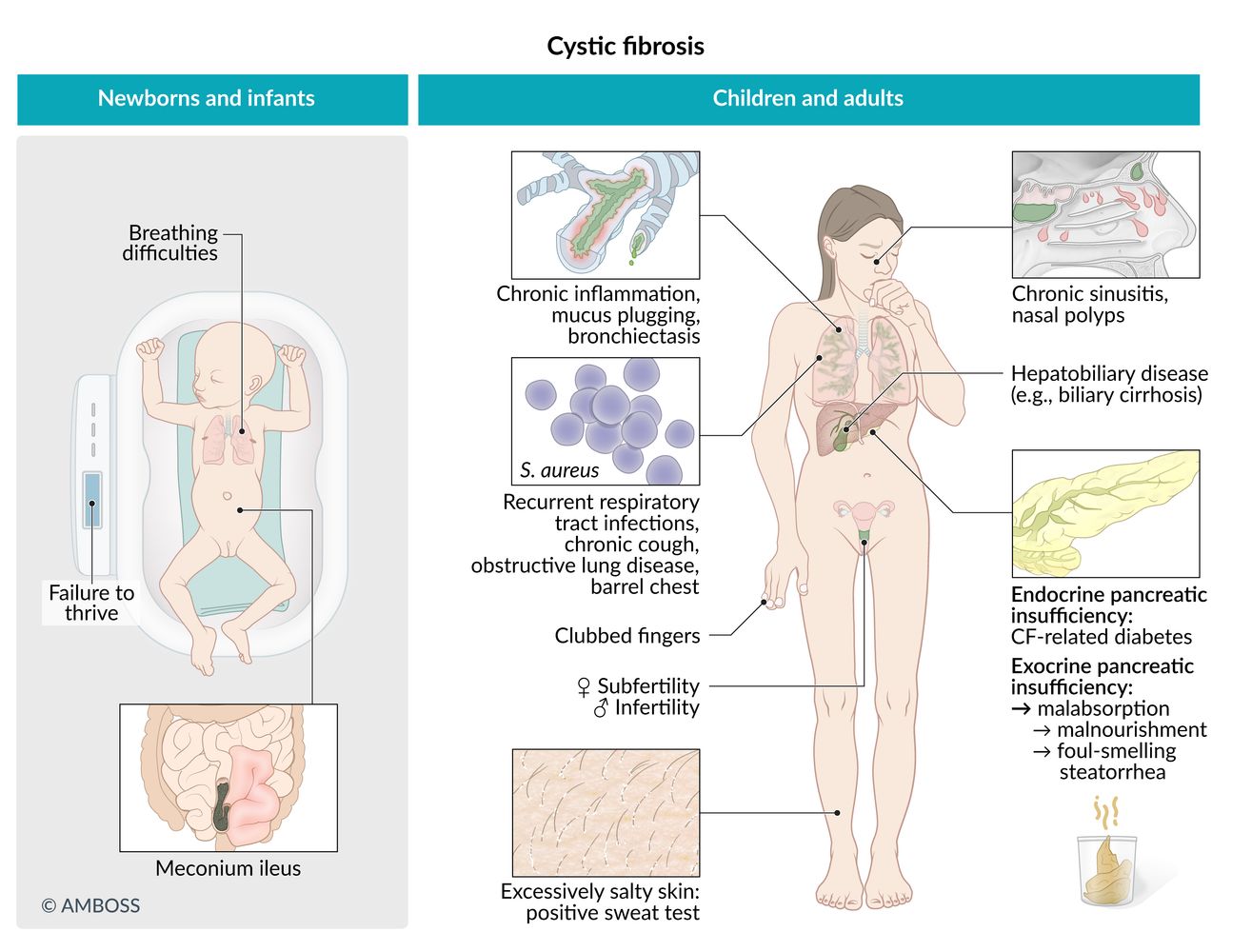
Gastrointestinal symptoms are common in children. The presence of these features during infancy should raise suspicion for CF.
- Meconium ileus (in newborns)
- Failure to thrive (due to malabsorption)
-
Pancreatic disease
- Pancreatitis
-
Exocrine pancreatic insufficiency
- Foul-smelling steatorrhea (fatty stools) may occur.
- Malabsorption
- Abdominal distention
- Diarrhea
- Hypoproteinemia
- Deficiency of fat-soluble vitamins
- CF-related diabetes mellitus (CFRD) [12]
-
Liver and bile duct abnormalities
- Cholecystolithiasis, cholestasis
- Fatty metamorphosis of the liver, eventually progressing to liver cirrhosis
- Biliary cirrhosis; with portal hypertension, jaundice, and/or esophageal varices
- Intestinal obstruction: abdominal distention, pain, and a palpable mass
- Rectal prolapse (rare)
In almost all cases of meconium ileus, cystic fibrosis is the underlying disease.

Respiratory [11]
Respiratory symptoms are common in adulthood. CF should be considered in individuals with the following features:
- Chronic obstructive lung disease with bronchiectasis
- Chronic sinusitis: nasal polyps may eventually develop
-
Recurrent or chronic productive cough and pulmonary infections
- S. aureus is the most common cause of recurrent pulmonary infection in infancy and childhood.
- P. aeruginosa is the most common cause of recurrent pulmonary infections in adulthood.
- Other commonly involved bacteria
- Burkholderia cepacia: can lead to cepacia syndrome, a severe necrotizing pneumonia that is often accompanied by rapid respiratory decline and can progress to sepsis [13]
- S. pneumoniae
- H. influenzae
- Increased susceptibility of individuals with CF to opportunistic, potentially life-threatening pathogens (e.g., Pseudomonas aeruginosa, Aspergillus)
- Infections with Pseudomonas aeruginosa → rapid decline in pulmonary function (patients with CF go through multiple antibiotic courses in their lifetime → increasing resistance of Pseudomonas aeruginosa to commonly used antibiotics) [14]
- A chronic infection with Aspergillus species may lead to allergic bronchopulmonary aspergillosis (see “Complications” below)
- Pulmonary obstruction and airway hyperreactivity may manifest with expiratory wheezing and/or dyspnea.
- Barrel chest , moist rales (indicate pneumonia), hyperresonance to percussion
- Hemoptysis
- Signs of chronic respiratory insufficiency: digital clubbing associated with chronic hypoxia

Sweat glands
- Particularly salty sweat
- Possible electrolyte wasting
Musculoskeletal
- Frequent fractures due to osteopenia
- Kyphoscoliosis
Urogenital
-
Urinary
- Nephrolithiasis, nephrocalcinosis
- Frequent urinary tract infections
-
Genital
-
Men: usually infertile
- Obstructive azoospermia; is common, spermatogenesis may be intact
- The vas deferens may be absent.
- Undescended testicle
-
Women: reduced fertility
- Viscous cervical mucus can obstruct fertilization.
- Menstrual abnormalities (e.g., amenorrhea)
- Delayed development of secondary sexual characteristics
-
Men: usually infertile
© AMBOSS
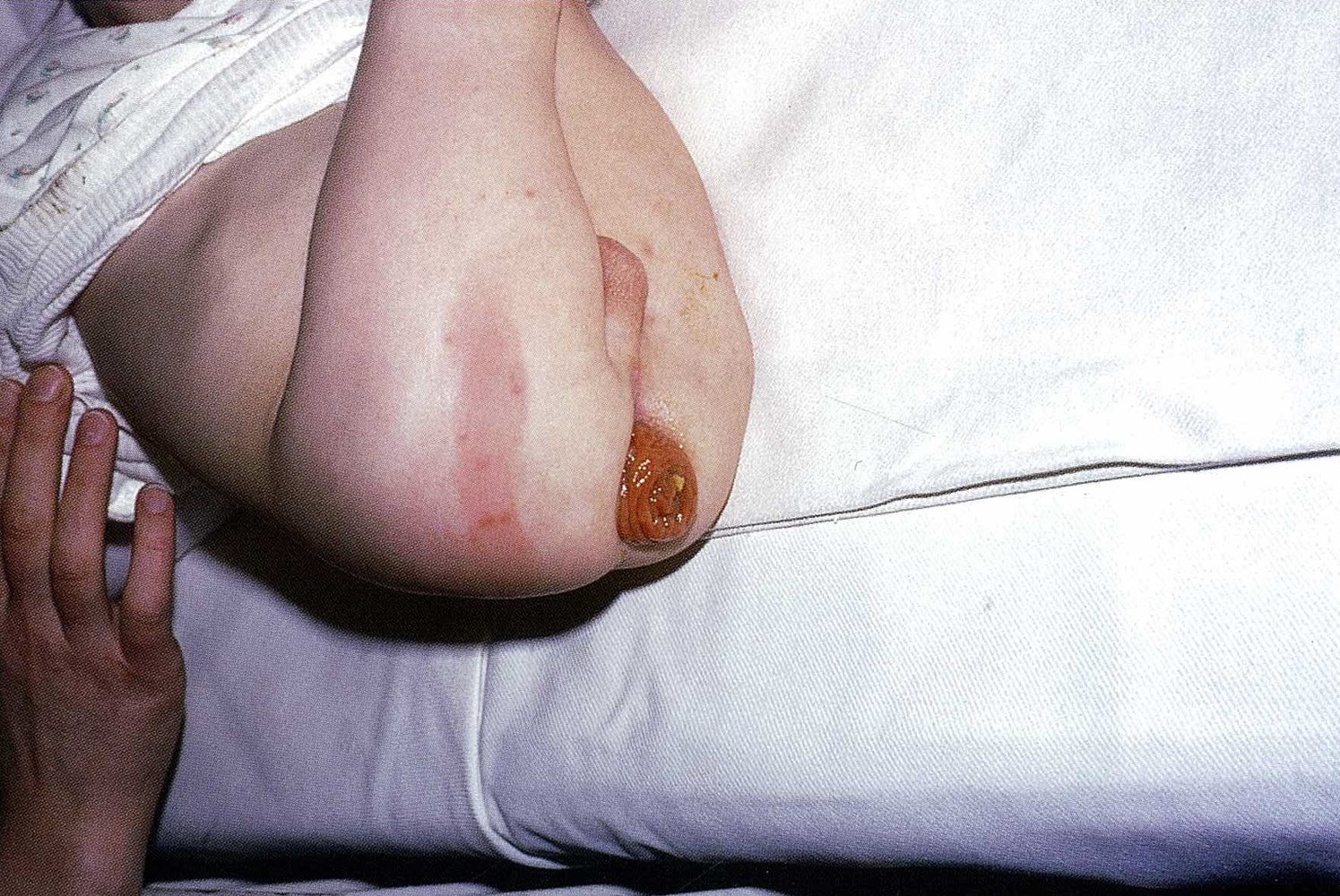
There is protrusion of all layers of the rectal wall through the external anal opening. Circular mucosal folds can be seen, which is consistent with a complete rectal prolapse.
Rectal prolapse in a child may be associated with a number of congenital conditions (e.g., cystic fibrosis or Hirschsprung disease).
Source: © IMPP
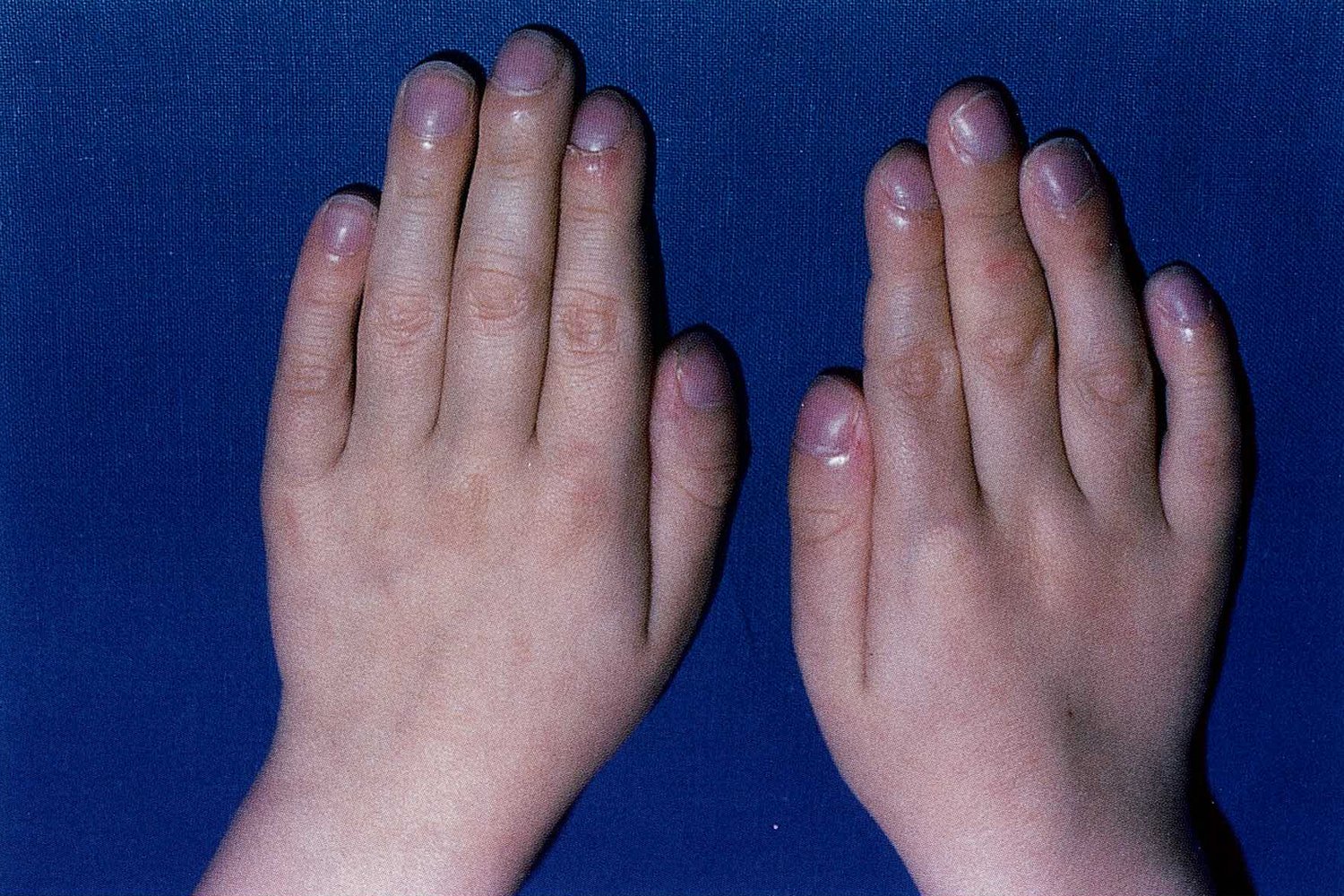
Enlarged, rounded nails with increased convexity (hippocratic nails) can be seen. There is club-shaped swelling of the distal phalanges in all fingers.
These findings indicate an underlying pathology causing chronic hypoxemia. Cystic fibrosis may manifest with chronic respiratory insufficiency.
Source: © IMPP
Newborn screening
Newborn screening (NBS) for CF is essential for early detection and treatment, which can improve health outcomes. In many countries, including the US, it is mandatory to screen for CF. [10][15]
- Sample collection: newborn blood spot test, performed via heel prick blood sampling in the first 24 and 48 hours of life
-
Screening tests [16]
-
Immunoreactive trypsinogen (IRT) : initial screening test
- An immunofluorescence assay that measures levels of IRT [17]
- Normal IRT levels: CF unlikely
- Elevated IRT levels: CF possible; additional screening tests required [16]
-
CFTR mutation testing (DNA testing): second-tier screening test
- Identifies the most common CF-causing CFTR mutations, including CF carriers [18]
- Cannot detect uncommon CFTR mutations that are not included on the panel.
-
Immunoreactive trypsinogen (IRT) : initial screening test
-
Screening protocols: a combination of CF screening methods to minimize false-positive and false-negative NBS results [18]
- IRT/DNA protocol: If initial IRT levels are elevated, a CFTR mutation test is performed on the same sample.
- IRT/IRT protocol: If initial IRT levels are elevated, a new sample is obtained and the test is repeated.
- Other protocols: e.g., IRT/IRT/DNA, IRT/DNA/full-gene analysis
- Next steps after positive NBS: Refer patients promptly to a specialist for a complete diagnostic workup and to start treatment if required.
Patients who test positive for CF during NBS should undergo a sweat test, ideally in the first 4 weeks of life. [10]
Patients with a presumptive diagnosis of CF (i.e., meconium ileus, positive NBS result with suggestive symptoms, or two CF-causing mutations detected during NBS) should immediately be referred to a specialized center and ideally evaluated within 24–72 hours. [15]
Diagnosis
Approach [15]
Diagnosis of CF is based on clinical findings and evidence of CFTR protein dysfunction.
- Presence of any of the following requires a follow-up with confirmatory testing:
- Positive newborn screening (NBS)
- First-degree family member with CF
- Typical clinical features of CF (e.g., chronic sinopulmonary disease, gastrointestinal and nutritional irregularities, syndromes of salt loss, obstructive azoospermia)
-
Any of the following findings are evidence of CFTR protein dysfunction:
- Sweat chloride testing with a chloride value ≥ 60 mmol/L
- Evidence of two CF-causing CFTR gene mutations and a sweat chloride test result ≥ 30 mmol/L
- Positive physiologic CFTR testing with abnormal nasal potential difference test or intestinal current measurement
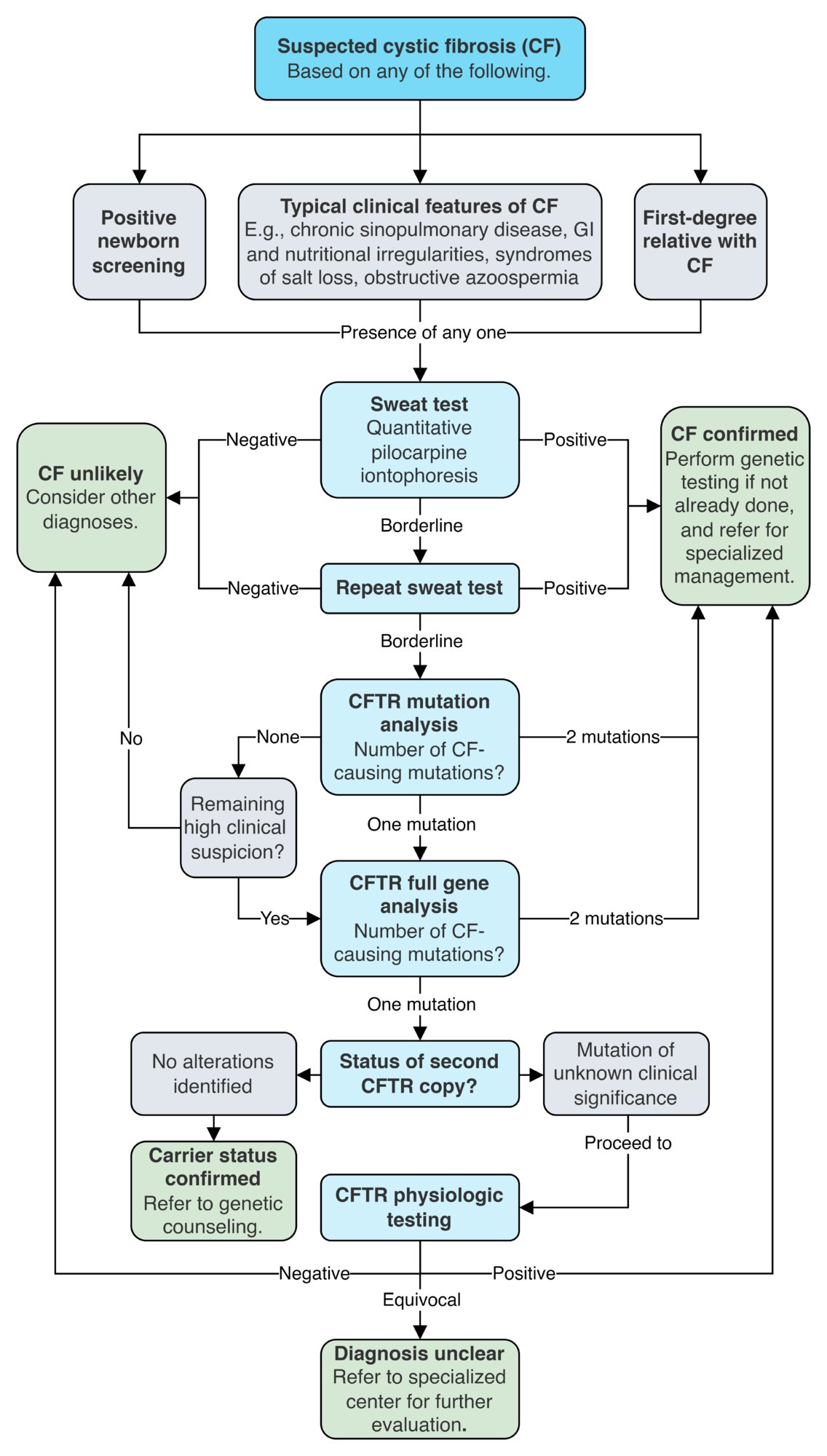
All patients with suspected CF should undergo a sweat test and CFTR genetic testing to help identify mutations that may affect management. [15]
| Confirmatory testing for CF [15][19] | ||||
|---|---|---|---|---|
| Order of testing | Results | Next steps | ||
| Sweat test (initial test) |
|
|
||
|
|
|||
|
|
|||
| CFTR mutation analysis |
|
|
||
|
|
|||
|
|
|||
| CFTR full gene analysis |
|
|
||
|
|
|||
|
|
|||
| CFTR physiologic testing |
|
|
||
|
|
|||
|
|
|||
Ideally, all patients with a confirmed diagnosis of CF should be evaluated at a specialized center (e.g., an accredited CF center) within 24–72 hours of diagnosis. [15]

Confirmatory tests
Sweat test (quantitative pilocarpine iontophoresis) [15]
- Indications: preferred initial test in all patients with suspected CF
- Method [10]
- Pilocarpine and an electrical current are applied to the skin to stimulate sweat production.
- Sweat is collected with absorbent pads and the Cl- concentration is measured.

CFTR dysfunction is confirmed with a positive sweat test (≥ 60 mmol/L). If the result is borderline (30–59 mmol/L), proceed to genetic testing and, if the diagnosis is still unclear, consider physiologic testing.
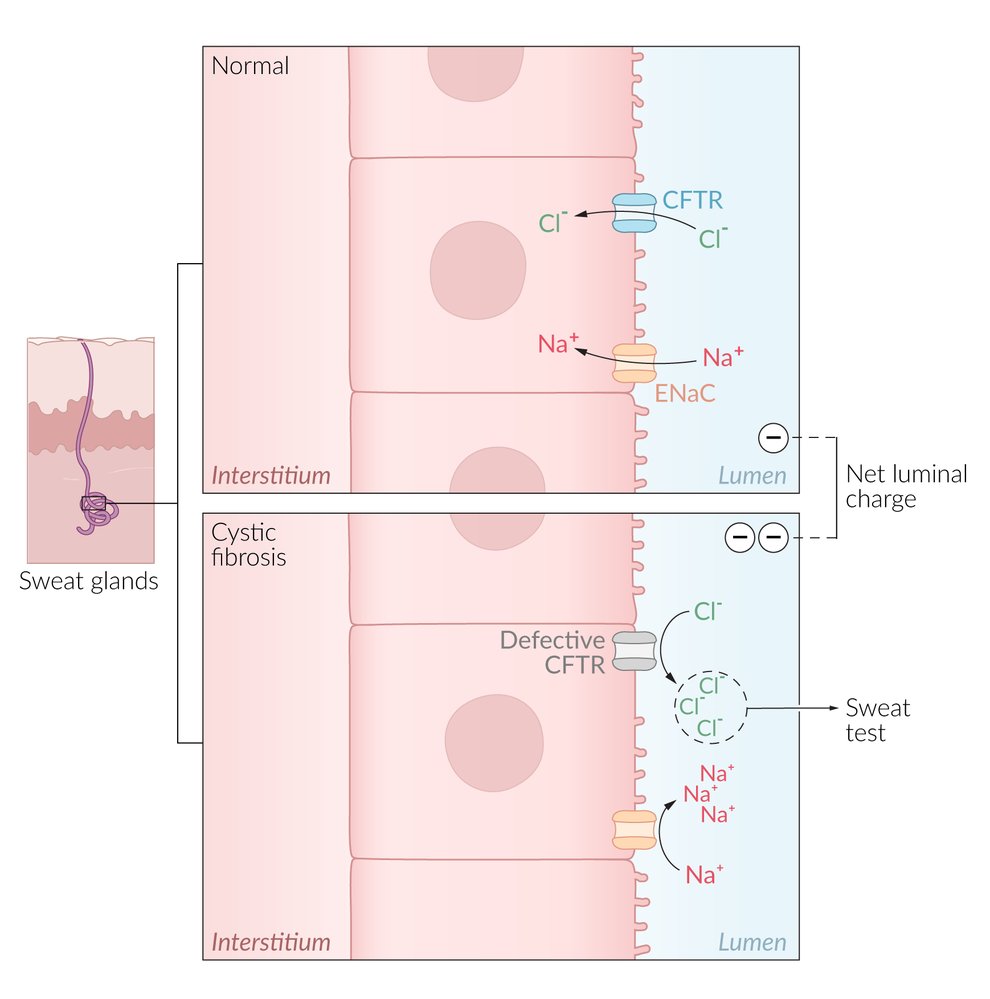
In most exocrine glands, intracellular Cl- is transported across the cell membrane into the lumen through the CFTR Cl- channel. In sweat glands, Cl- is transported in the opposite direction, from the lumen into the cell. In CF, a defect in the CFTR Cl- channel results in an accumulation of Cl- and, subsequently, of Na+ in the lumen of the sweat gland, leading to an increased concentration of NaCl in the sweat.
CFTR genetic testing [15][20][21]
-
Indication
- Carrier screening
- Confirmatory test after equivocal sweat test
- Guidance of targeted therapy (e.g., CFTR modulators)
-
Modalities
- CFTR mutation analysis: only tests for the most common CF-causing CFTR mutations
- CFTR full gene analysis: includes gene sequencing and deletion/duplication analysis to detect less common CFTR mutations
CFTR physiologic testing [15][22][23]
-
Procedure
- Exposing specific tissues to different standardized solutions results in predictable ion movements and voltage changes.
- Ionic charges and voltage responses can be measured and compared with standard reference ranges for patients with and without CF.
-
Modalities
-
Nasal potential difference test
- In vivo assessment of CFTR function in the respiratory epithelium, performed in the nasal cavity
- Results include e.g., more negative baseline potential difference and no difference in nasal potential difference after administration of a chloride-free solution
- Intestinal current measurement: ex vivo assessment of CFTR function in the intestinal epithelium, performed in a fresh rectal biopsy sample
-
Nasal potential difference test
Additional investigations
The pulmonary status of all patients with CF should be assessed at the time of diagnosis using pulmonary function tests (PFTs) and imaging, in addition to microbiological studies to detect respiratory pathogens. These studies should be repeated regularly to monitor for disease progression, which allows for early initiation of any required interventions.
-
Pulmonary function tests: indicated at diagnosis and repeated every 3 months for monitoring
- Modalities: predominantly spirometry in patients ≥ 6 years of age [24]
- Findings: obstructive pattern, e.g., ↓ FEV1:FVC ratio (see “Obstructive lung diseases”) [25][26][20]
-
Imaging: chest x-ray or chest CT are indicated at baseline and every 2–4 years in patients with stable lung function
- Early findings: may be normal or show subtle signs of air trapping and hyperinflation
-
Late findings
- Signs of obstructive lung disease (e.g., air trapping, hyperinflation)
- Reticular nodular pattern
- Bronchiectasis
-
Microbiology: findings can help prevent and treat exacerbations [20][27]
- Bacterial sputum cultures most commonly show growth of some of the following pathogens:
- Gram positive: Staphylococcus aureus
- Gram negative: Hemophilus influenzae, Pseudomonas aeruginosa (a defining pathogen in CF), Burkholderia cepacia , Achromobacter spp., and Stenotrophomonas maltophilia
- Additional studies include: sputum smear and culture for acid-fast bacilli, fungal cultures
- Bacterial sputum cultures most commonly show growth of some of the following pathogens:
Monitoring with sputum cultures helps guide the selection of antibiotics to prevent and treat exacerbations. [20][27]
Pseudomonas, Stenotrophomonas, and/or Burkholderia infections are associated with rapid lung function deterioration and higher rates of lung transplantation. [27]
-
Additional screening and monitoring
- Routine laboratory studies
- CBC: may show anemia; potentially leukocytosis during exacerbations
- BMP: findings are variable, e.g., contraction alkalosis and hypokalemia
- Nutritional assessment
- Weight, height, and head circumference
- Blood tests: total protein and albumin (typically low), fat-soluble vitamins
- Stool tests: low levels of pancreatic elastase suggest exocrine pancreatic insufficiency
- Liver chemistries and coagulation panel: to assess liver function and screen for CF-related liver disease (CFLD)
- Fasting glucose: as a part of the screening for CF-related diabetes (CFRD)
- Audiology exam
- CT head may show opacification of sinuses in patients with features of chronic sinusitis
- Routine laboratory studies
Sweat losses of NaCl and H2O lead to contraction of the ECF volume and RAAS activation (similar to the effects of loop diuretics). This can result in increased renal reabsorption of NaCl and H2O and excretion of H+ and K+, causing alkalosis and hypokalemia.


© AMBOSS
Sweating is induced using pilocarpine and an electrical current, and collected using preweighed filter paper covered with plastic wrap (to prevent evaporation) for up to 30 minutes. The concentration of chloride is measured and compared to reference ranges. A chloride concentration of ≥ 60 mmol/L reflects a likely diagnosis of cystic fibrosis.
© AMBOSS
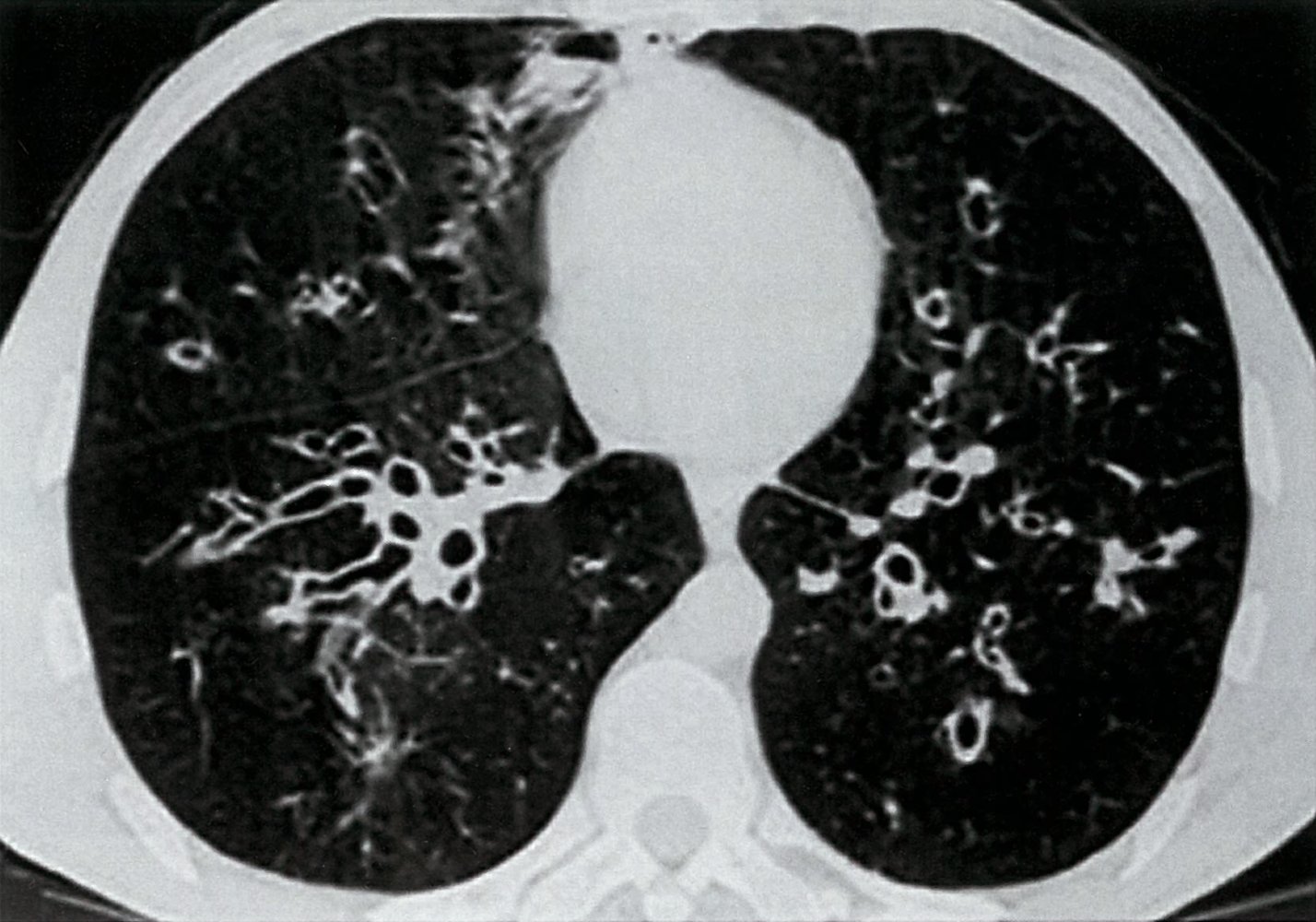
CT chest (axial section)
Numerous bilateral dilated bronchi (bronchiectasis) show wall thickening and an increased bronchoarterial ratio. Several bronchi imaged in cross-section have a diameter greater than the accompanying pulmonary artery diameter (1; signet ring sign), while others imaged in long axis have parallel walls (2; tram track sign).
Source: © IMPP
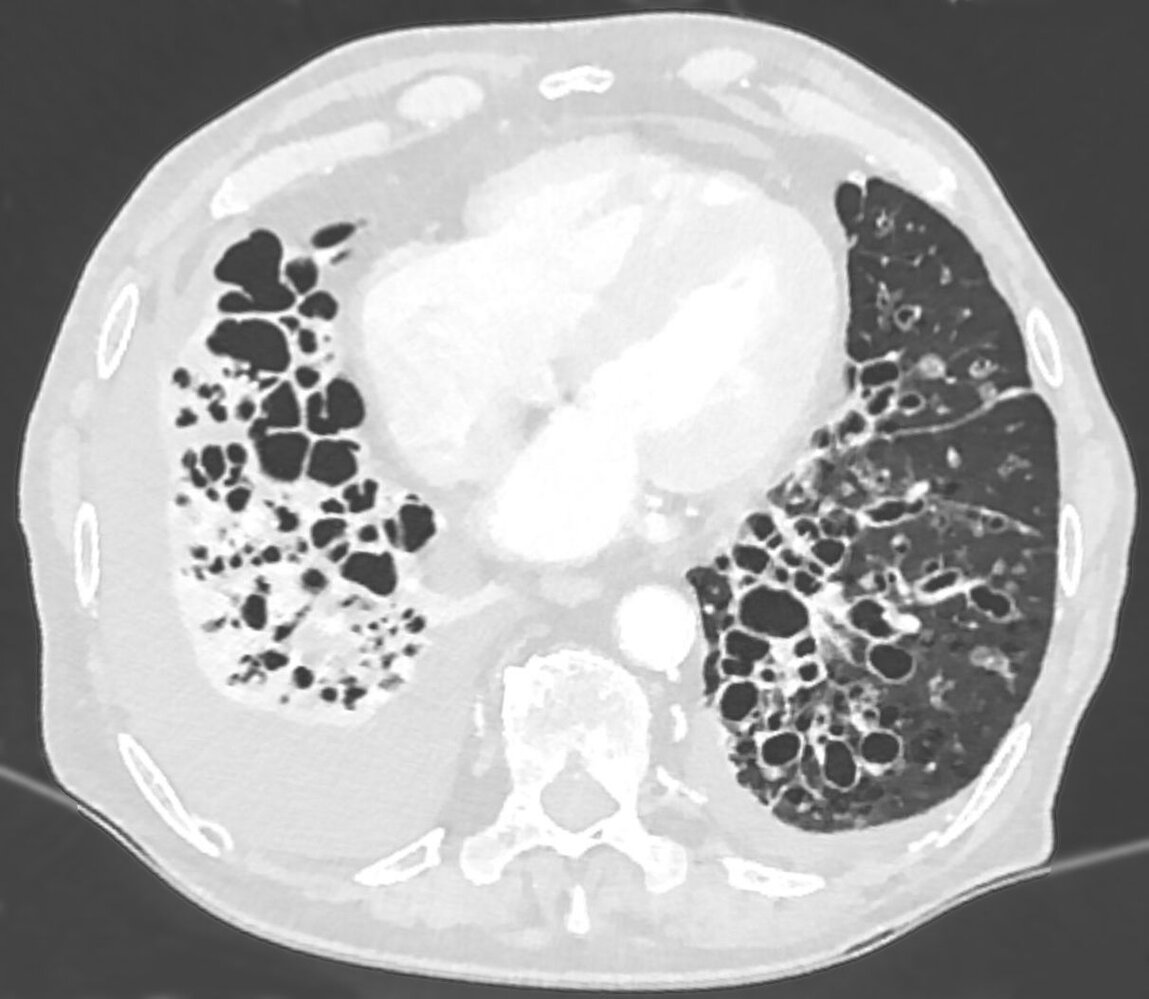
CT chest (axial plane; lung window)
Multiple dilated bronchi in the lower lobes and lingula are accompanied by inflammatory wall thickening. Many bronchi are larger than the adjacent pulmonary arteries (signet ring sign; examples indicated by red arrowheads), in contrast to the normal appearance of approximately equal size. Additional abnormalities include right lower lobe consolidation with volume loss and bilateral pleural effusions.
The severe form of bronchiectasis seen here is termed cystic bronchiectasis.
Source: “Massive Bronchiektasen - CT LF axial 001.jpg” by Hellerhoff, Wikimedia Commons, licensed under CC BY-SA 3.0. The supplementary image with overlays of relevant areas was adapted from the image mentioned above and licensed under CC BY-SA 3.0.
{kind=link}
Differential diagnoses
- Primary ciliary dyskinesia (PCD)
- Allergic bronchopulmonary aspergillosis (ABPA)
- Congenital immunodeficiency disorders
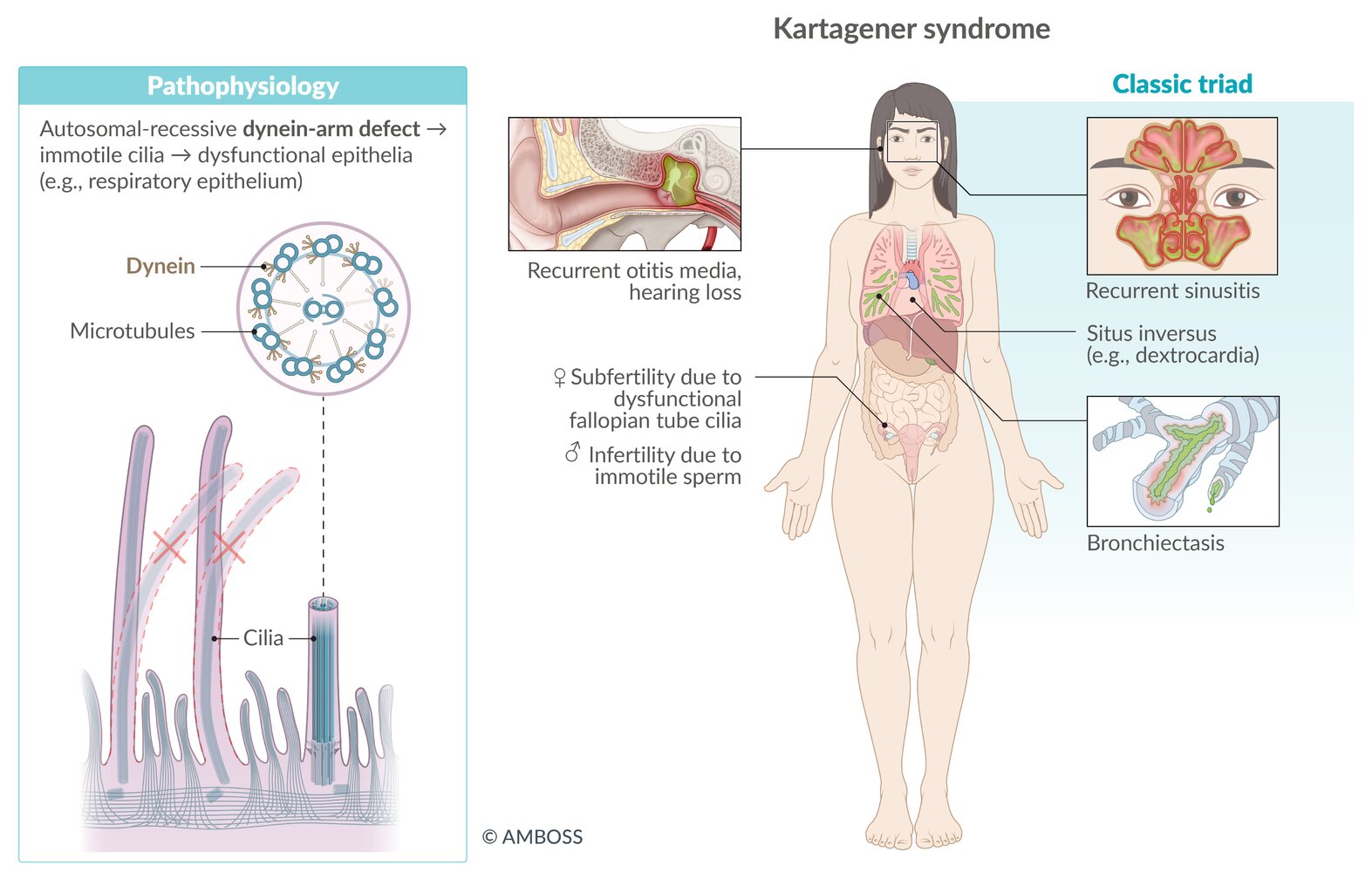
Primary ciliary dyskinesia [28][29]
- Definition: rare autosomal recessive disorder characterized by absent or dysmotile cilia caused by a defect in the dynein arm of microtubules
-
Clinical features
- Chronic productive cough
- Recurrent otitis, sinusitis, and nasal polyps
- Bronchiectasis
- Conductive hearing loss
- Displaced heart sounds (as a result of dextrocardia)
- Infertility in men due to decreased sperm motility as a result of defective flagella
- Reduced fertility in women (and rarely ectopic pregnancy) due to defective cilia in fallopian tubes
- Kartagener syndrome: classic triad of situs inversus, recurrent sinusitis, and bronchiectasis
-
Diagnostics
- Nasal nitric oxide test: reduced nasal nitric oxide (screening test)
- Genetic tests for dynein arm mutations
- Chest x-ray: bronchiectasis, dextrocardia, and situs inversus (suggests Kartagener syndrome)
- Electron microscopy: abnormal cilia
- Treatment: depends on individual clinical presentation and course
You can memorize the cause of Kartagener syndrome by thinking of Kartagener's restaurant that only has 'take-out' service because there is no dine-in (dynein).
Kartagener syndrome is a subtype of primary ciliary dyskinesia characterized by the triad of situs inversus, chronic sinusitis, and bronchiectasis.

")


The differential diagnoses listed here are not exhaustive.
© AMBOSS
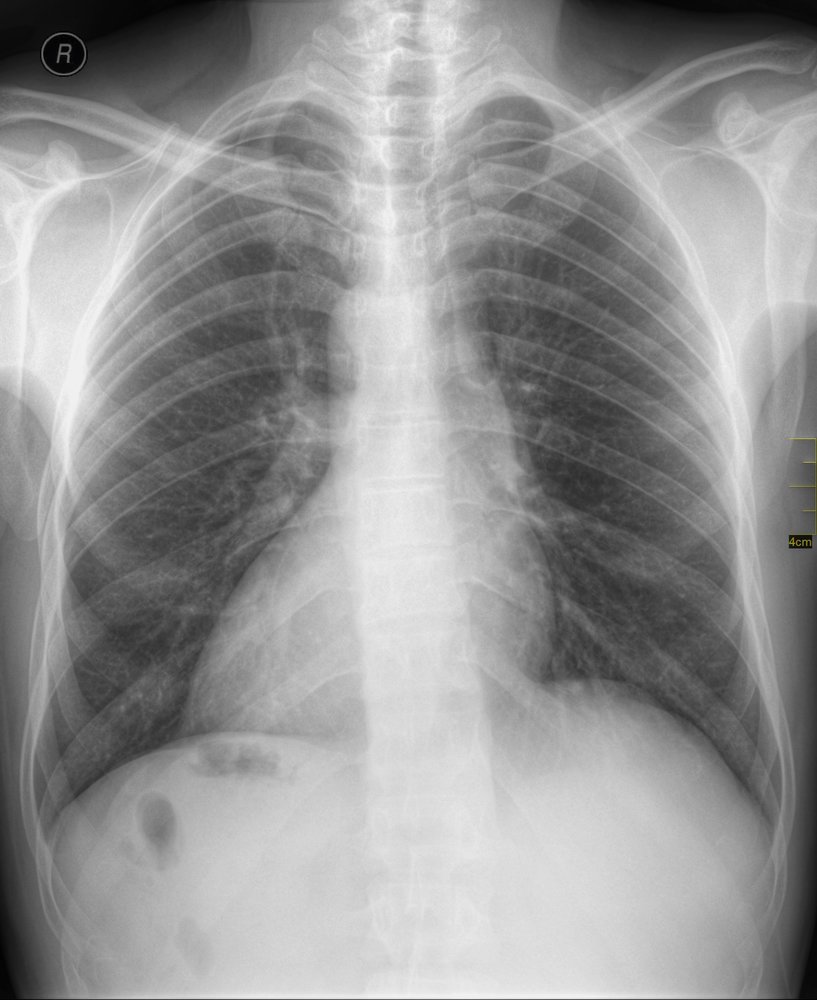
X-ray chest (PA view)
The aortic arch (dotted line), cardiac apex (arrow), and gastric bubble (red overlay) are seen on the right side of the body. Opacification in the left upper abdominal quadrant (green overlay) is the result of a left-sided liver.
Source: “Situs inversus chest Nevit” by Nevit, Wikimedia Commons, licensed under CC BY 3.0. The supplementary image with overlays of relevant areas was adapted from the image mentioned above (© AMBOSS).
{kind=link}
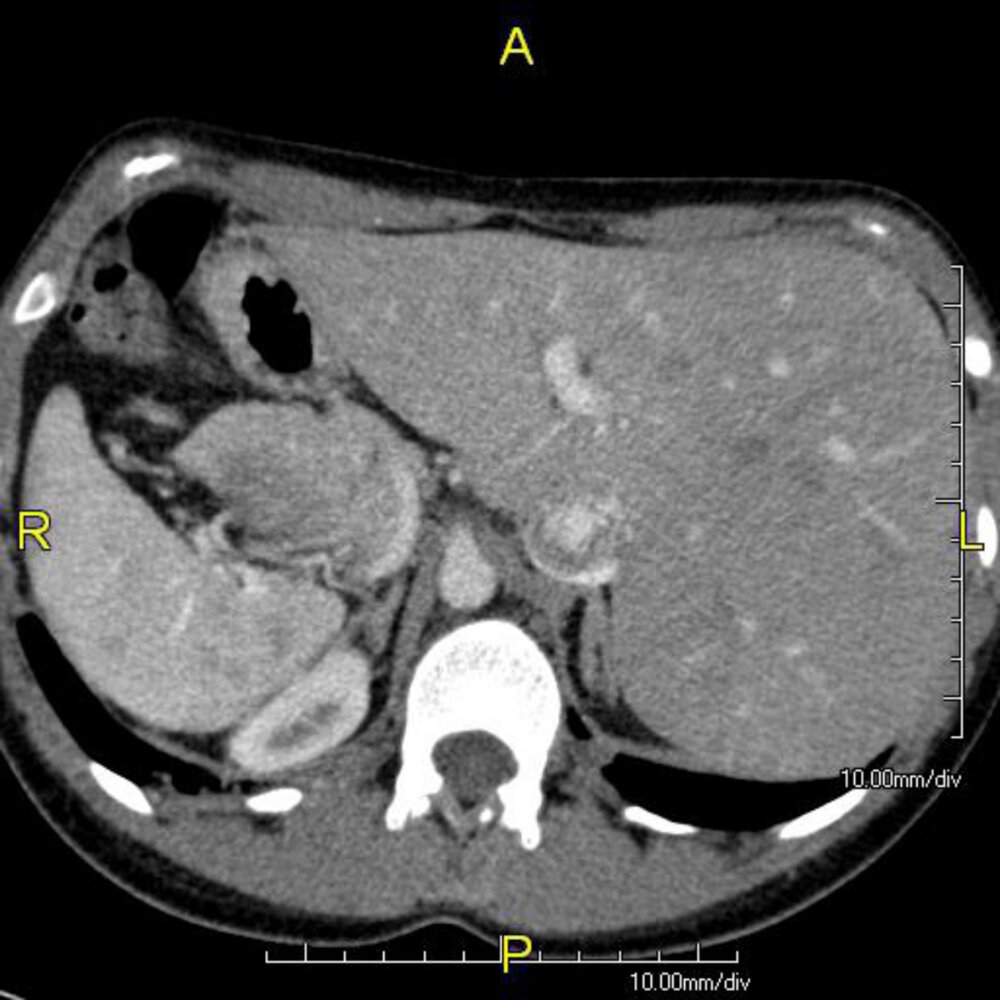
CT abdomen (with contrast; axial plane)
Mirror-image reversal of abdominal organs (situs inversus) is present, with the liver (L) located on the patient's left and the spleen (S) on the right.
Situs inversus is one of the clinical features seen in Kartagener syndrome.
K: kidney
This image is an adaptation. Source of original image: Wikimedia Commons. Original title: “Primary ciliary dyskinesia”. Created by: John S. To, MD. Licensed under Public Domain. Modifications to original image: - upscaled.
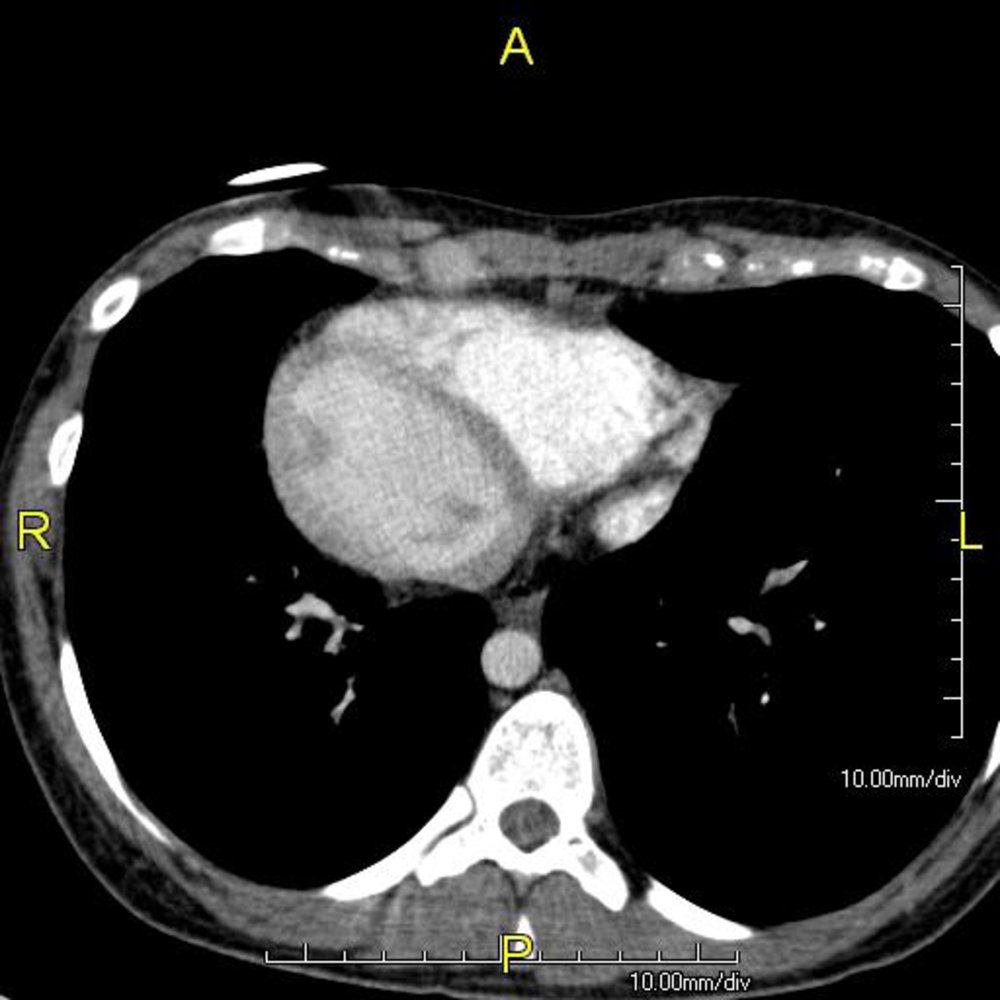
CT chest (with contrast; axial plane)
The base-to-apex cardiac axis is directed to the patient's right (dextrocardia). The right ventricle (RV) and left ventricle (LV) are reversed. This patient also presented with right to left reversal of the morphologic atria and abdominal organs (situs inversus; not shown).
Dextrocardia with situs inversus is part of the triad of manifestations seen in Kartagener syndrome, which also includes chronic sinusitis and bronchiectasis.
Source: "Primary ciliary dyskinesia", John S. To, MD, Wikimedia Commons licensed under Public Domain
Management
General principles [20][30]
- All patients with CF require periodic follow-up with a multidisciplinary team for specialized management.
- Management should include the following goal-directed interventions:
- Preservation of lung function
- Pharmacological and nonpharmacological interventions
- Prevention of infection and reduction of exacerbations
- Optimization of nutrition
- Screening and monitoring for comorbidities and complications
- Preservation of lung function
- Patients with certain mutations may benefit from treatment with CFTR modulators.
- Acute pulmonary exacerbations require rapid and effective treatment.
Preservation of lung function [20][31][32]
Lung function is preserved by combining pharmacological and nonpharmacological interventions to improve mucociliary clearance, reduce mucus viscosity, and mobilize secretions.
Pharmacological interventions [20][31]
-
High-dose ibuprofen: can slow the progression of lung disease
- Indication: Consider in patients 6–17 years of age with mild disease (FEV1 > 60% of predicted).
- Avoid in patients with a history of hemoptysis.
-
Bronchodilators: SABA (e.g., albuterol), LABA (e.g., salmeterol)
- SABA indications: prior to inhaled therapies and/or chest physiotherapy , patients with episodes of airway hyperresponsiveness
- Long-term use is not routinely recommended [20][31]
-
Mucolytics [20][31][32]
- Hypertonic saline nebulization; (6–7% NaCl): mucociliary and osmotic effect; that can improve mucociliary clearance and thin the mucus [32]
- Dornase alfa , aerosolized: a recombinant DNase that thins the mucus by breaking down extracellular DNA in sputum [32]
- Other therapies
- N-acetylcysteine: efficacy is unproven in CF [20][31]
- Corticosteroids (inhaled and systemic): insufficient evidence; not routinely used [32]
Nonpharmacological interventions
-
Airway clearance techniques: a mainstay of CF treatment that loosens and mobilizes mucus secretions
- Conventional chest physiotherapy (CPT): postural drainage with percussion and/or clapping
- Alternative airway clearance methods
- High-frequency chest compression
- Airway oscillating devices
- Positive expiratory pressure devices
- Huff coughing
- Exercise (e.g., swimming, jogging, cycling)
Patients with CF benefit from a regular airway clearance routine that combines pharmacological measures with airway clearance techniques to preserve lung function and reduce symptoms. [20][31]
An airway clearance session generally begins with SABA therapy to open the airways, followed by mucolytics to thin the mucus, then airway clearance techniques. [20][31]
Prevention of infection and reduction of exacerbations in CF [20][27][31][32][33]
- Pulmonary exacerbations in patients with CF are often triggered by chronic lung infections of pathogenic organisms.
- Eradication and/or suppression regimens can prevent exacerbations and improve lung function.
- Consider treatment after early detection of relevant pathogens during routine surveillance sputum cultures.
- Eradication regimens include:
- P. aeruginosa: inhaled tobramycin
- MRSA: inhaled vancomycin PLUS oral antibiotics , PLUS extrapulmonary eradication
- Suppression regimens
- Indicated in patients with chronic infections
- May suppress the bacterial load, improve lung function, and/or prevent exacerbations
- Azithromycin: may be given long term, with benefits relying on its antiinflammatory effect [20][27][31][32]
Antibiotic eradication and suppression regimens for patients with CF should always be selected in consultation with a specialist.
Optimization of nutrition [34]
There is a direct correlation between BMI and pulmonary lung function in patients with CF. The following are common dietary recommendations for patients with CF:
- High-energy diet to compensate for increased demand (target BMI ≥ 50thpercentile) [20][35]
- Pancreatic enzyme supplements [10]
- Additional NaCl intake [35]
- Oral supplementation of fat-soluble vitamins (ADEK) [20]
CFTR modulators [30][36]
A relatively new therapy for the long-term management of CF that targets specific defects in the CFTR protein to improve its function.
-
Indications
- Approved for patients with specific CFTR mutations (e.g., ΔF508, G511D mutations)
- Their use can potentially reduce CF complications and comorbidities
-
Mechanism of action: improve CFTR protein function by targeting underlying protein defects [31]
- Potentiators (e.g., ivacaftor): increase CFTR Cl- channel gate opening and conductance and improve Cl- transport
- Correctors (e.g., lumacaftor, tezacaftor, elexacaftor): improve protein folding, protein stability, and the transport of functional CFTR protein to the cell surface [37]
-
Combination therapy
- Two or more CFTR modulators with different mechanisms of action can be combined to synergistically improve CFTR protein quantity and function.
- Combination therapy increases the yield of CFTR modulators and can therefore benefit larger numbers of patients. [10]
Perform CFTR genetic testing in all patients diagnosed with CF to assess for specific mutations that can be targeted by CFTR modulators.
CFTR modulators can improve FEV1, help patients gain weight, reduce exacerbations, and decrease sweat Cl- concentration. [36]
The names of CFTR modulators include all the letters C-F-T-R: IvaCaFToR, LumaCaFToR, TezaCaFToR, and ElexaCaFToR.
Additional measures [20][38]
-
Oxygen therapy: frequent oxygen monitoring and support for chronic respiratory insufficiency as needed
- Intermittent oxygen: indicated for desaturation during exercise and sleep
- Long-term oxygen: indicated for hypoxia on room air and borderline hypoxia with evidence of cor pulmonale
-
Double-lung transplant
- A treatment option for patients with end-stage lung disease [20][38]
- Common indications for transplantation referral include:
- Low (< 30% of predicted) or rapidly declining FEV1
- Evidence of pulmonary hypertension
- Significant clinical decline.
- Provide emotional support and recommend additional mental health support to help patients manage their chronic condition and end-of-life care decisions. [32]
- Discuss the patient's long-term goals of care.
CF is a chronic, progressive, and ultimately life-shortening condition. Preferably, CF should be managed by a multidisciplinary team that can provide comprehensive care, which emphasizes shared decision-making.
Acute pulmonary exacerbations
General principles [8][20][27]
-
Increased severity or new onset of clinical symptoms can be a sign of acute pulmonary exacerbation.
- There are no universally accepted diagnostic criteria.
- Findings are varied and include increased dyspnea or cough, changes in sputum production, weight loss, and fever
- Start culture-guided antibiotic therapy for pulmonary exacerbations.
- Additional measures
- Optimize nutrition.
- Increase airway clearance.
- Provide respiratory support as needed; see “Oxygen therapy” for details.
- Glucocorticoids: insufficient evidence, not routinely used
- Order inpatient contact precautions.
Treat pulmonary exacerbations quickly and aggressively to prevent an irreversible decline in lung function. [32]
Diagnostics [8][20][27]
Obtain a detailed clinical history, including the frequency and severity of respiratory symptoms. Patients may report weight loss, fever, and/or hemoptysis. Perform a thorough physical examination, paying particular attention to new auscultation findings and abnormal vital signs (e.g., increased respiratory rate, low oxygen saturation).
- Laboratory: CBC, BMP, liver chemistries, and consider blood gas
- Assess pulmonary status.
- PFTs: possible decline in FEV1
- Chest x-ray: may show new infiltrates
- Obtain new sputum cultures upon admission.

Antibiotics [27]
Antibiotic therapy in patients with CF is challenging because of a high prevalence of antibiotic resistance following frequent antibiotic use and the presence of multiple pathogenic organisms. Involve specialists early.
Choose initial antibiotics based on the speciation and sensitivities of the patient's recent routine sputum cultures. Adjust the antibiotics, if needed, once the results from the new sputum culture have been reported.
| Antibiotic therapy for CF pulmonary exacerbations [27] | |
|---|---|
| Pathogen | Example regimens |
| Staphylococcus aureus |
|
| Pseudomonas aeruginosa |
|
| Other pathogens |
|
Disposition [8][20][27]
- Outpatient management: Specialists may provide ambulatory management for mild exacerbations with inhaled and/or oral antibiotics.
- Indications for inpatient management [8][20]
- Resistant bacteria with no PO antibiotic options
- Exacerbations with a poor response to outpatient management
- Moderate to severe disease or complicating comorbidities
- Critical care: Consider especially for young patients with a good baseline status and transplantation candidates.
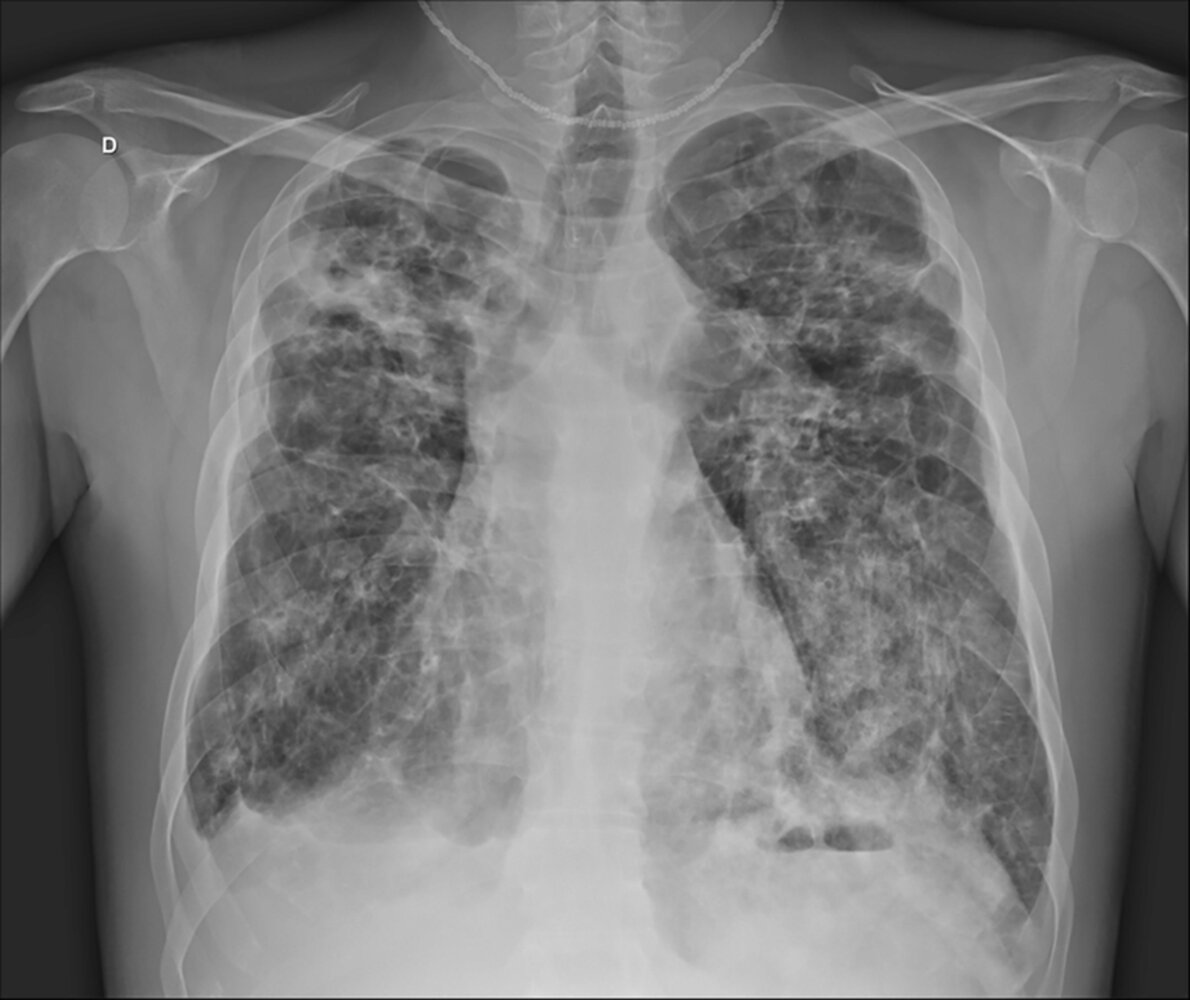
X-ray chest (PA view) of a patient with cystic fibrosis and pneumonia
Cystic airspaces (examples indicated by green overlay) and thick-walled dilated bronchi (bronchiectasis; examples indicated by red overlay) can be seen throughout both lungs. The pulmonary hila are elevated from the normal position as a result of chronic bilateral upper lobe volume loss. Additionally, the well-seen left hilum is enlarged (indicated by arrows), reflecting the presence of pulmonary hypertension.
Bilateral ill-defined lung opacities in this patient represent concurrent pneumonia, which was accompanied by a right pleural effusion (dashed line).
Source: “Figure1, in: Influenza A non-H1N1 associated with acute respiratory failure and acute renal failure in a previously vaccinated cystic fibrosis patient” by Louise Piva Penteado, Cecília Susin Osório, Antônio Balbinotto, Paulo de Tarso Roth Dalcin, Revista Brasileira de Terapia Intensiva, licensed under CC BY 4.0. The supplementary image with overlays of relevant areas was adapted from the image mentioned above (© AMBOSS).
Complications
Gastrointestinal
Gastrointestinal complications of CF include:
-
Meconium ileus: failure to pass the first stool in neonates (meconium usually passes within the first 24–48 hours after birth)
- Etiology: Cystic fibrosis is the cause in > 90% of cases.
- Clinical findings: signs of a distal small bowel obstruction (thick meconium plugs the distal ileum)
- Bilious vomiting
- Abdominal distention
- No passing of meconium or stool
- Diagnostics
-
X-ray abdomen (with contrast agent) ; [40]
- Dilated small bowel loops
- Microcolon: narrow caliber of the colon, as it is still unused (meconium has not been passed through yet)
- Neuhauser sign (soap bubble appearance): a mottled or bubble-like appearance in the distal ileum and/or cecum as a result of meconium mixing with swallowed air [41]
- Air-fluid levels are uncommon because of the viscous consistency of meconium.
-
X-ray abdomen (with contrast agent) ; [40]
-
Differential diagnosis: See “Differential diagnosis of intestinal obstruction in neonates.”
- Treatment
- Enema with a contrast agent
- Surgery is required in complicated cases (e.g., intestinal perforation, volvulus)
- Small bowel obstruction: can also occur in older children and adults
-
Distal intestinal obstruction syndrome (DIOS) : blockage of the small intestines by thickened stool in the distal ileum and right colon [42]
- Highest prevalence in the second and third decades of life
- Tends to occur in individuals with pancreatic insufficiency
- Clinical manifestations include abdominal pain and distention, a palpable cecal mass, and flatulence.


")
Respiratory
- Allergic bronchopulmonary aspergillosis (ABPA): ∼ 10% of individuals with CF develop this condition. [43][44]
-
Pulmonary emphysema
- Pneumothorax
- Cor pulmonale
We list the most important complications. The selection is not exhaustive.
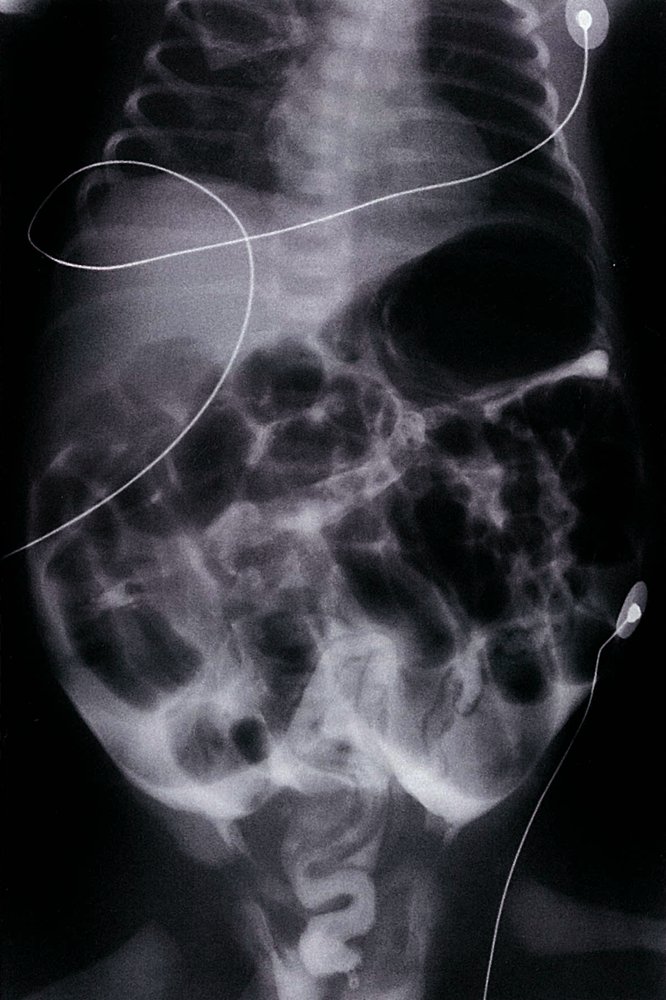
Barium enema (AP view) of a neonate with meconium ileus
Multiple dilated gas-filled loops of small bowel are visible and opaque barium contrast outlines a microcolon (sections of microcolon indicated by green outlines). The findings are consistent with meconium ileus.
Contrast is also seen in multiple locations within the peritoneal space (examples indicated by red overlay) as a result of bowel perforation.
Blue overlay: ECG electrodes
Source: © IMPP
© AMBOSS
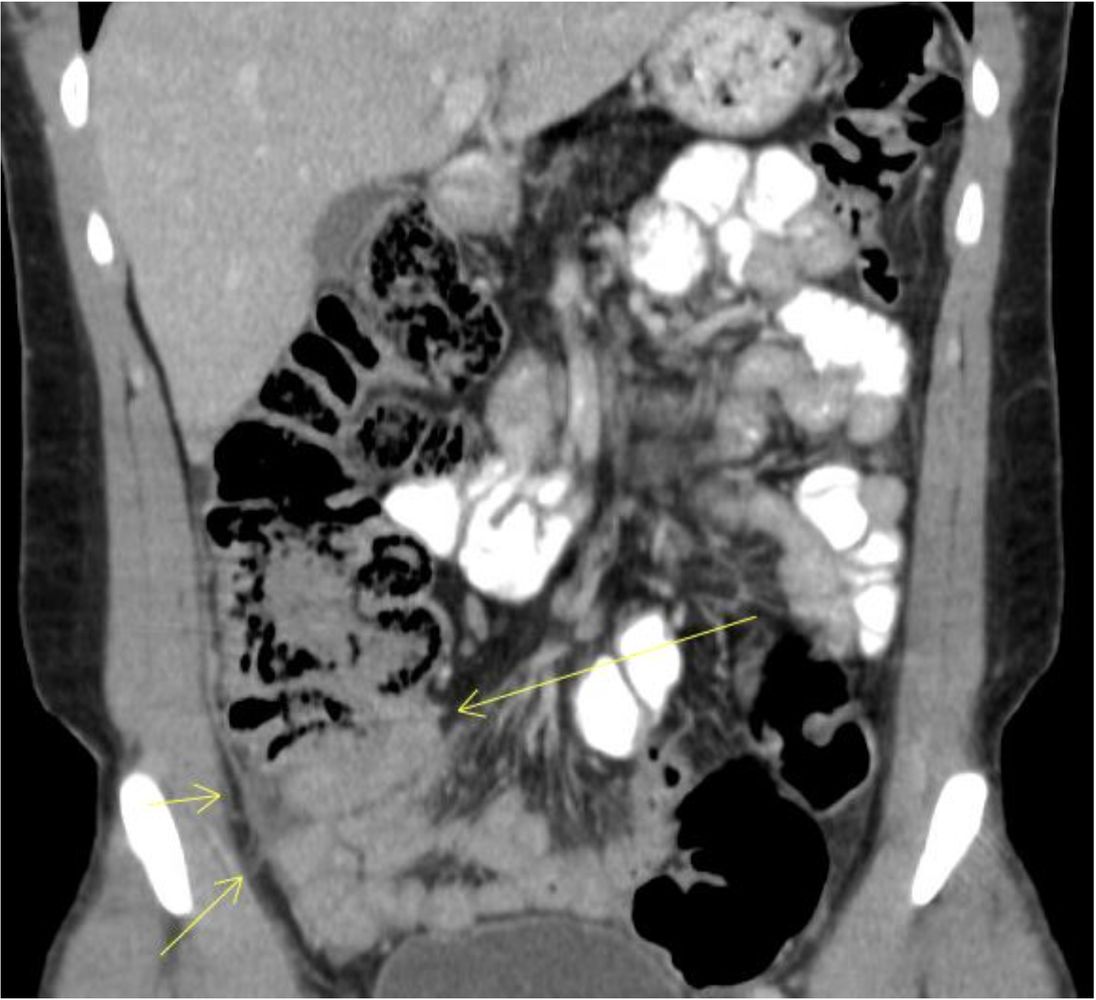
CT abdomen (with IV and oral contrast; coronal plane) of a patient with cystic fibrosis
Thickened bowel is visible in the right lower quadrant in the region of the terminal ileum and appendix (arrows). This finding can be seen in DIOS. Adjacent inflammatory changes are also present on this image, with increased density of fatty and fascial tissue.
Source: “Fig. 2, in: Acute Appendicitis Masquerading Distal Intestinal Obstruction Syndrome in Adult Cystic Fibrosis” by Nanavati SM, Patel H, Melki G, et al., Case Reports in Gastrointestinal Medicine, licensed under CC BY 4.0. Modifications: cropped.
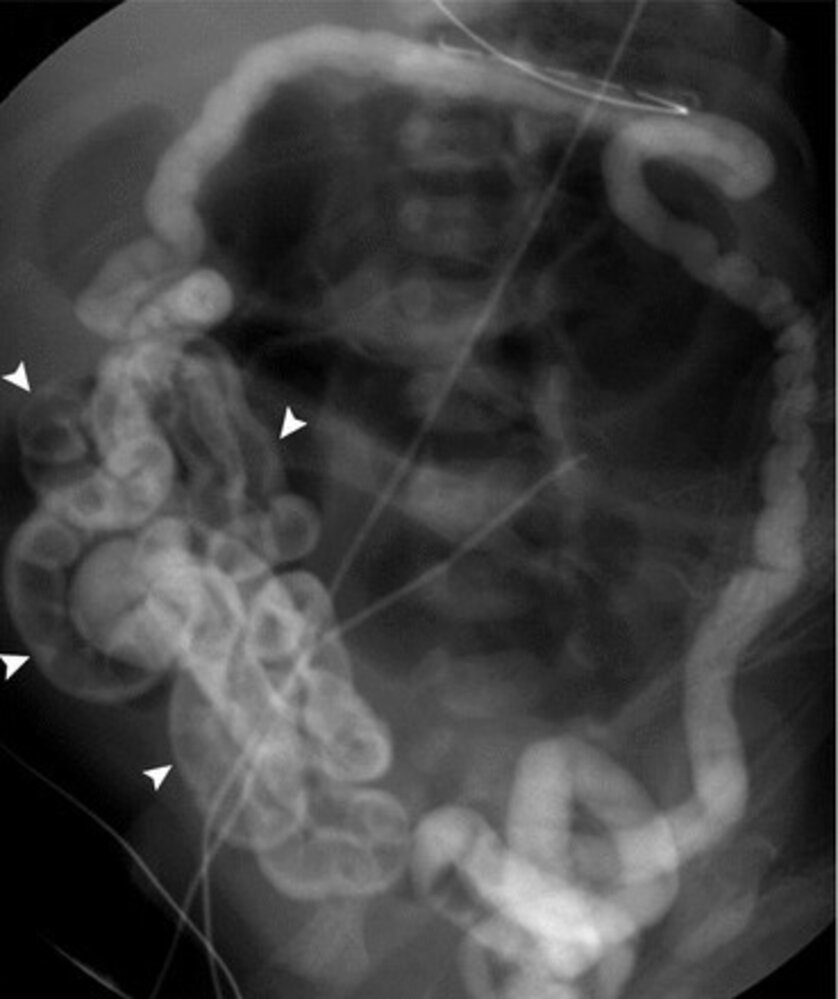
Fluoroscopy (barium enema; AP view)
Contrast opacifies a microcolon and has refluxed into the ileum, outlining numerous pellets of impacted meconium (indicated by overlay). Dilated gas-filled loops of small bowel are present proximal to the obstruction in the mid abdomen.
Source: “The Pediatric Gastrointestinal Tract: What Every Radiologist Needs to Know” by Emily A. Dunn, Øystein E. Olsen, and Thierry A. G. M. Huisman, Diseases of the Abdomen and Pelvis 2018-2021: Diagnostic Imaging, licensed under CC BY 4.0. Modifications: letter removed. The supplementary image with overlays of relevant areas was adapted from the image mentioned above (© AMBOSS).
Prognosis
- Median life expectancy: 39 years [45]
- Individuals with CF who have pancreatic sufficiency tend to present with mainly pulmonary symptoms in late childhood/early adulthood and generally have a milder course of disease [46]
- The main determinant of life expectancy is the severity of pulmonary disease: chronic respiratory infections and mucus plugging → bronchiectasis (irreversible) → progressive respiratory failure → death
Prevention
- Annual influenza vaccine for all affected individuals > 6 months with inactivated influenza vaccine
- Pneumococcal vaccine (see ”Immunization schedule”)
- Palivizumab: antibody against respiratory syncytial virus (RSV) for infants < 24 months
- Long-term treatment with azithromycin may be used to prevent recurrent pulmonary infections.
External Resources
References
- Navarro S. "Recopilación histórica de la fibrosis quística". Gastroenterol Hepatol. 39(1). :36-42. (2016)
- Bosch L, Bosch B, De Boeck K, et al. "Cystic fibrosis carriership and tuberculosis: hints toward an evolutionary selective advantage based on data from the Brazilian territory.". BMC Infect Dis. 17(1). :340. (2017)
- Sibley CD, Rabin H, Surette MG. "Cystic fibrosis: a polymicrobial infectious disease". Future Microbiol. 1(1). :53-61. (2006)
- "The molecular genetics of cystic fibrosis". https://www.who.int/genomics/publications/reports/en/. [2004-01-01]
- Maitra R, Hamilton J. "Altered biogenesis of ΔF508-CFTR following treatment with Doxorubicin". Cell Physiol Biochem. 20(5). :465-472. (2007)
- Ratjen FA. "Cystic fibrosis: pathogenesis and future treatment strategies.". Respir Care. 54(5). :595-605. (2009)
- Tsui L-C, Dorfman R. "The cystic fibrosis gene: a molecular genetic perspective". Cold Spring Harb Perspect Med. 3(2). :a009472-a009472. (2013)
- "Cystic Fibrosis: Does CFTR Malfunction Alter pH Regulation?". http://www.intechopen.com/books/genetic-disorders/cystic-fibrosis-does-cftr-malfunction-alter-ph-regulation-. [2013-01-09]
- Moran A, Dunitz J, Nathan B, et al. "Cystic fibrosis-related diabetes: current trends in prevalence, incidence, and mortality". Diabetes Care. 32(9). :1626-1631. (2009)
- Jones AM, Dodd ME, Webb AK. "Burkholderia cepacia: current clinical issues, environmental controversies and ethical dilemmas.". The European respiratory journal. 17(2). :295-301. (2001)
- Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL. "Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis". Pediatr Pulmonol. 34(2). :91-100. (2002)
- Farrell PM, White TB, Ren CL, et al. "Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation". J Pediatr. 181. :S4-S15.e1. (2017)
- Farrell PM, Rosenstein BJ, White TB, et al. "Guidelines for Diagnosis of Cystic Fibrosis in Newborns through Older Adults: Cystic Fibrosis Foundation Consensus Report". J Pediatr. 153(2). :S4-S14. (2008)
- Dickinson KM, Collaco JM. "Cystic Fibrosis". Pediatr Rev. 42(2). :55-67. (2021)
- Yankaskas JR, Marshall BC, Sufian B, Simon RH, Rodman D. "Cystic Fibrosis Adult Care". Chest. 125(1). :1S-39S. (2004)
- Girardet A, Viart V, Plaza S, et al. "The improvement of the best practice guidelines for preimplantation genetic diagnosis of cystic fibrosis: toward an international consensus". Eur J Hum Genet. 24(4). :469-478. (2015)
- Minso R, Schulz A, Dopfer C, et al. "Intestinal current measurement and nasal potential difference to make a diagnosis of cases with inconclusive CFTR genetics and sweat test". BMJ Open Resp Res. 7(1). :e000736. (2020)
- Schüler D, Sermet-Gaudelus I, Wilschanski M, et al. "Basic protocol for transepithelial nasal potential difference measurements". J Cyst Fibros. 3. :151-155. (2004)
- Davis SD, Ratjen F, Brumback LC, et al. "Infant lung function tests as endpoints in the ISIS multicenter clinical trial in cystic fibrosis". J Cyst Fibros. 15(3). :386-391. (2016)
- Vilozni D, Bentur L, Efrati O, et al. "Spirometry in Early Childhood in Cystic Fibrosis Patients". Chest. 131(2). :356-361. (2007)
- Lombardi E, Gambazza S, Pradal U, Braggion C. "Lung clearance index in subjects with cystic fibrosis in Italy". Ital J Pediatr. 45(1). (2019)
- Chmiel JF, Aksamit TR, Chotirmall SH, et al. "Antibiotic management of lung infections in cystic fibrosis. I. The microbiome, methicillin-resistant Staphylococcus aureus, gram-negative bacteria, and multiple infections.". AnnalsATS. 11(7). :1120-9. (2014)
- Moran A, Brunzell C, Cohen RC, et al. "Clinical Care Guidelines for Cystic Fibrosis-Related Diabetes: A position statement of the American Diabetes Association and a clinical practice guideline of the Cystic Fibrosis Foundation, endorsed by the Pediatric Endocrine Society". Diabetes Care. 33(12). :2697-2708. (2010)
- Flume PA, Mogayzel PJ, Robinson KA, et al. "Cystic Fibrosis Pulmonary Guidelines". Am J Respir Crit Care Med. 180(9). :802-808. (2009)
- Lucas JS, Burgess A, Mitchison HM, et al. "Diagnosis and management of primary ciliary dyskinesia". Arch Dis Child. 99(9). :850-856. (2014)
- Shapiro AJ, Davis SD, Polineni D, et al. "Diagnosis of Primary Ciliary Dyskinesia. An Official American Thoracic Society Clinical Practice Guideline". Am J Respir Crit Care Med. 197(12). :e24-e39. (2018)
- "Meconium ileus". https://radiopaedia.org/articles/meconium-ileus. [2016-02-16]
- "Neuhauser Sign (Distal Ileum)". https://radiopaedia.org/articles/neuhauser-sign-distal-ileum. [2018-01-01]
- Fields TM, Michel SJ, Butler CL, Kriss VM, Albers SL. "Abdominal Manifestations of Cystic Fibrosis in Older Children and Adults". American Journal of Roentgenology. 187(5). :1199-1203. (2006)
- Laufer P, Fink JN, Bruns WT, et al. "Allergic bronchopulmonary aspergillosis in cystic fibrosis.". J Allergy Clin Immunol. 73(1 Pt 1). :44-8. (1984)
- Stevens DA, Moss RB, Kurup VP, et al. "Allergic bronchopulmonary aspergillosis in cystic fibrosis--state of the art: Cystic Fibrosis Foundation Consensus Conference.". Clin Infect Dis. 37 Suppl 3. :S225-64. (2003)
- MacKenzie T, Gifford AH, Sabadosa KA, et al. "Longevity of patients with cystic fibrosis in 2000 to 2010 and beyond: Survival analysis of the Cystic Fibrosis Foundation Patient Registry". Ann Intern Med. 161(4). :233. (2014)
- Mount DB. "Clinical manifestations and treatment of hypokalemia in adults". UpToDate. UpToDate. https://www.uptodate.com/contents/clinical-manifestations-and-treatment-of-hypokalemia-in-adults?source=search_result&search=hypokalemia&selectedTitle=1~150#H3819731. [2016-01-07]
- Ren CL et al. "Cystic Fibrosis Foundation Pulmonary Guidelines. Use of Cystic Fibrosis Transmembrane Conductance Regulator Modulator Therapy in Patients with Cystic Fibrosis". Annals of the American Thoracic Society. 15(3). :271-280. (2018)
- Mogayzel PJ, Naureckas ET, Robinson KA, et al. "Cystic Fibrosis Pulmonary Guidelines". Am J Respir Crit Care Med. 187(7). :680-689. (2013)
- Cohen-Cymberknoh M, Shoseyov D, Kerem E. "Managing Cystic Fibrosis". Am J Respir Crit Care Med. 183(11). :1463-1471. (2011)
- Mogayzel PJ, Naureckas ET, Robinson KA, et al. "Cystic Fibrosis Foundation Pulmonary Guideline*. Pharmacologic Approaches to Prevention and Eradication of InitialPseudomonas aeruginosaInfection". Ann Am Thorac Soc. 11(10). :1640-1650. (2014)
- Stallings VA, Stark LJ, Robinson KA, Feranchak AP, Quinton H. "Evidence-Based Practice Recommendations for Nutrition-Related Management of Children and Adults with Cystic Fibrosis and Pancreatic Insufficiency: Results of a Systematic Review". J Am Diet Assoc. 108(5). :832-839. (2008)
- Turck D, Braegger CP, Colombo C, et al. "ESPEN-ESPGHAN-ECFS guidelines on nutrition care for infants, children, and adults with cystic fibrosis". Clin Nutr. 35(3). :557-577. (2016)
- Ridley K, Condren M. "Elexacaftor-Tezacaftor-Ivacaftor: The First Triple-Combination Cystic Fibrosis Transmembrane Conductance Regulator Modulating Therapy". J Pediatr Pharmacol Ther. 25(3). :192-197. (2020)
- Lopes-Pacheco M. "CFTR Modulators: Shedding Light on Precision Medicine for Cystic Fibrosis". Front Pharmacol. 7. (2016)
- Kapnadak SG, Dimango E, Hadjiliadis D, et al. "Cystic Fibrosis Foundation consensus guidelines for the care of individuals with advanced cystic fibrosis lung disease". J Cyst Fibros. 19(3). :344-354. (2020)
- Gilbert, DN; Chambers, HF. "Sanford Guide to Antimicrobial Therapy 2020". Antimicrobial Therapy, Inc. (2020). ISBN: 9781944272135
- Ross LF. "Newborn Screening for Cystic Fibrosis: A Lesson in Public Health Disparities". J Pediatr. 153(3). :308-313. (2008)
- Therrell BL, Hannon WH, Hoffman G, Ojodu J, Farrell PM. "Immunoreactive trypsinogen (IRT) as a biomarker for cystic fibrosis: Challenges in newborn dried blood spot screening". Mol Genet Metab. 106(1). :1-6. (2012)
- Bender LM, Cotten SW, Willis MS. "Kids in America: Newborn Screening for Cystic Fibrosis". Lab Med. 42(10). :595-601. (2011)