Summary
Bleeding disorders are a group of heterogeneous conditions characterized by defects in hemostasis that lead to an increased susceptibility to bleeding (also known as hemorrhagic diathesis). They are classified into disorders of primary hemostasis (when caused by a platelet abnormality), disorders of secondary hemostasis (when caused by defects in the extrinsic and/or intrinsic pathway of the coagulation cascade), and hyperfibrinolysis (when there is increased clot degradation). Although clinical features may overlap, mucocutaneous bleeding (e.g., epistaxis, petechiae, gastrointestinal bleeding) is associated with disorders of primary hemostasis, and bleeding into potential spaces (e.g., hemarthrosis, muscular bleeding) is characteristic of disorders of secondary hemostasis. The diagnostic workup of a bleeding disorder begins with a detailed clinical assessment, the CBC, and a coagulation panel. This typically allows the disorder to be classified as one of primary or secondary hemostasis. Specialized studies are then required to determine the specific etiology so that treatment can be initiated. Treatment may include transfusion of blood products, replacement of specific coagulation factors, or administration of adjuvant medications (e.g., tranexamic acid or desmopressin).
Hemostasis
Overview [1][2]
Hemostasis is the physiological process by which a bleeding stops. Its final result is a thrombus (blood clot), which consists of blood cells and fibrin strands. Hemostasis involves the following mechanisms:
-
Primary hemostasis
- Vascular hemostasis: transient vasoconstriction and vWF activation following endothelial injury
- Platelet hemostasis: adhesion, activation, and aggregation of platelets, which results in the formation of a platelet plug (white thrombus)
- Secondary hemostasis: activation of the coagulation cascade, which results in the formation of a fibrin clot (red thrombus)
Primary hemostasis
- Definition: : processes involved in the formation of a platelet plug (white thrombus) following endothelial injury
-
Vascular hemostasis
-
Endothelial injury results in:
-
Neural stimulation reflexes and endothelin release → transient vasoconstriction, leading to:
- Reduced blood flow
- Platelet accumulation at the vessel walls
- Exposure of subendothelial collagen → circulating von Willebrand factor binds to the exposed collagen
-
Neural stimulation reflexes and endothelin release → transient vasoconstriction, leading to:
-
Von Willebrand factor (vWF): plasma protein that is synthesized by and stored in endothelial cells (in Weibel-Palade bodies) and platelets (in α-granules)
- Mediates platelet adhesion and aggregation
- Binds factor VIII (and thereby prevents its degradation)
-
Endothelial injury results in:
-
Platelet hemostasis
-
Platelet adhesion: platelets bind to vWF via platelet GpIb receptor at the endothelial injury site
- Ristocetin normally activates vWF to bind to glycoprotein Ib
-
Platelet activation: After binding to vWF, platelets change their shape and release mediators that lead to activation of more platelets (positive feedback). ; These mediators include:
- Adenosine diphosphate (ADP): promotes adhesion of platelets to endothelium
- Thromboxane A2 (TXA2): activates additional platelets and promotes vasoconstriction
- Calcium: required for secondary hemostasis
- Platelet-activating factor (PAF): a phospholipid mediator that is produced by platelets and inflammatory cells (e.g., neutrophils, monocytes, macrophages), involved in platelet aggregation and activation and local inflammatory response
-
Platelet aggregation
- Mediated by GpIIb/IIIa-receptor and fibrinogen → formation of a white thrombus composed of platelets and fibrinogen
- A white thrombus is transient, unstable, and easily dislodged. It stabilizes through the process of secondary hemostasis.
-
Platelet adhesion: platelets bind to vWF via platelet GpIb receptor at the endothelial injury site




Secondary hemostasis
-
Definition: : processes that lead to stabilization of the platelet plug; (white thrombus; ) by creating a fibrin network
- Coagulation cascade: a sequence of events triggered by the activation of the intrinsic or extrinsic pathway of coagulation that results in the formation of a stable thrombus
-
Coagulation factors
- Substances that interact with each other to promote blood coagulation
- Activated factors are designated with an “a” (e.g., activated factor VII = factor VIIa).
-
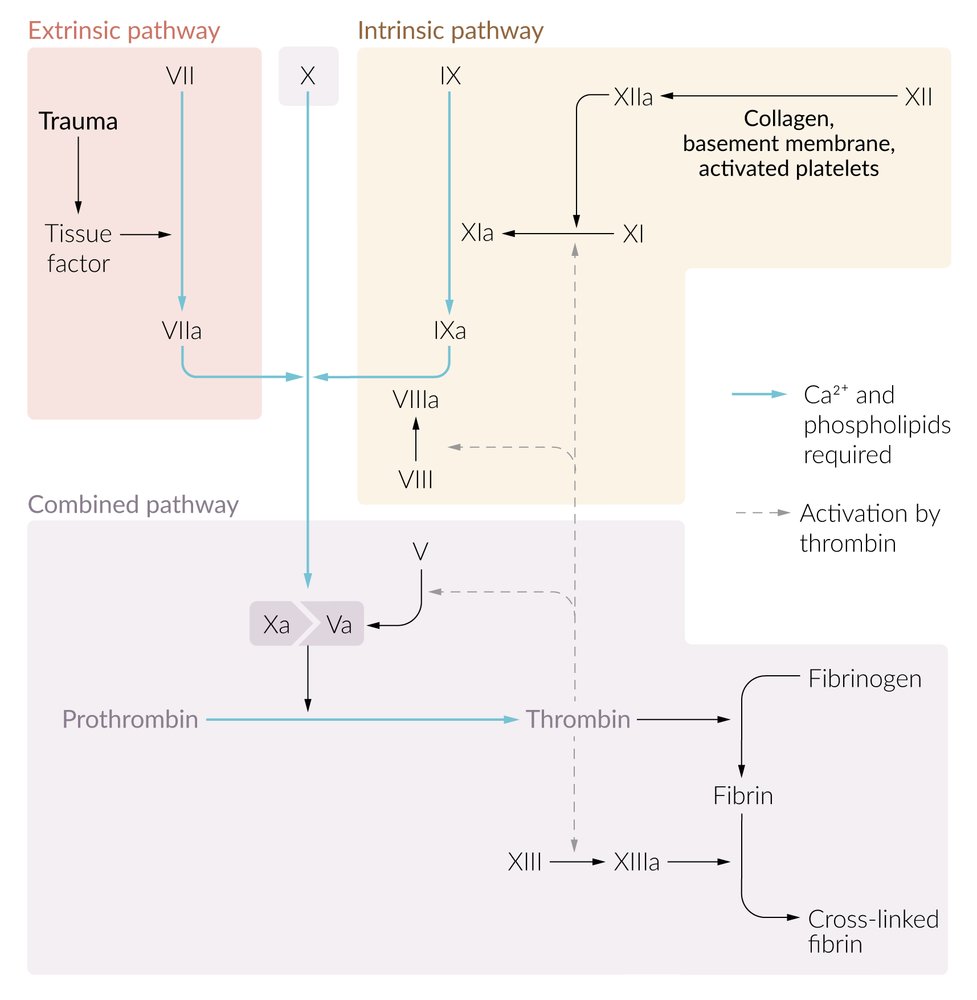
Extrinsic pathway of coagulation: triggered by endothelial injury
-
Tissue factor (factor III) activates factor VII.
- Tissue factor is expressed on the surface of subendothelial muscle cells and fibroblasts.
- Factor VII: vitamin K-dependent coagulation factor produced by the liver
- Factor VIIa and tissue factor form a complex (TF-FVIIa). This step requires calcium (factor IV) found on the surface of fibrocytes and activated platelets.
- TF-FVIIa activates factor X and factor IX.
-
Tissue factor (factor III) activates factor VII.
-
Intrinsic pathway of coagulation
-
Exposed collagen, kallikrein, and kininogen (HMWK) activate factor XII.
- Factor XII (Hageman factor): coagulation factor that also plays a role in inflammatory response by activating the kallikrein system, which leads to the production of bradykinin
- Factor XIIa activates factor XI.
- Thrombin activates factor XI and factor VIII.
- Factor XIa activates factor IX.
- Factors VIIIa and IXa form a complex (mediated by calcium) that activates factor X.
- This causes a positive feedback loop of factor X and thrombin activation via the intrinsic pathway.
-
Exposed collagen, kallikrein, and kininogen (HMWK) activate factor XII.
-
Common pathway of coagulation: The extrinsic and intrinsic pathway both end in the common pathway.
- Factor Xa and factor Va form a complex (mediated by calcium) that cleaves prothrombin (factor II) to thrombin (factor IIa).
- Thrombin cleaves fibrinogen (factor I) into insoluble fibrin (factor Ia) monomers.
- Crosslinks of the fibrin network are stabilized by factor XIIIa; → formation of a fibrin network → fibrin closely binds to the platelet plug, forming a stable thrombus (secondary thrombus or red thrombus)
| Overview of coagulation factors | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Factor number | Descriptive name | Activated by | Involvement in pathways | Function | |||||||
| Common | Intrinsic | Extrinsic | |||||||||
| I† |
|
|
✓ |
|
|||||||
| II |
|
|
✓ | ✓ | ✓ |
|
|||||
| III† |
|
|
✓ |
|
|||||||
| IV† |
|
|
✓ | ✓ | ✓ |
|
|||||
| V* |
|
|
✓ |
|
|||||||
| VII* |
|
|
✓ |
|
|||||||
| VIII* |
|
|
✓ |
|
|||||||
| IX* |
|
|
✓ | ||||||||
| X* |
|
|
✓ |
|
|||||||
| XI* |
|
|
✓ |
|
|||||||
| XII* |
|
|
✓ |
|
|||||||
| XIII* |
|
|
✓ |
|
|||||||
| * = preferred term [3] † = rarely used term | |||||||||||
The coagulation cascade requires the presence of calcium ions (factor IV).
A helpful way of remembering the coagulation factors of the extrinsic pathway is 3 + 7 = 10: Tissue factor (factor III) and factor VII form a complex that activates factor X of the common pathway.
A helpful way of remembering the coagulation factors of the common pathway is 10/5 = 2 × 1: Factors Xa and Va form a complex that cleaves prothrombin (factor II) to thrombin (IIa). Factor IIa then cleaves fibrinogen (I) into insoluble fibrin monomers (Ia).




Inhibition of hemostasis
In order to prevent hypercoagulability as well as excessive bleeding, activation of the coagulation cascade and the processes that inhibit it occur simultaneously in the circulatory system (procoagulant-anticoagulant balance).
- Tissue factor pathway inhibitor: inhibits tissue factor
-
Protein C and protein S: Activated protein C and its cofactor protein S form the activated protein-C complex (APC complex), which inhibits factors Va and VIIIa.
- Vitamin K-dependent synthesis in the liver
- Shorter half-life than vitamin K-dependent coagulation factors (relevant for treatment with vitamin K antagonists, e.g., warfarin)
-
Clinical relevance
- APC resistance
- Factor V Leiden
- Protein C deficiency, protein S deficiency
-
Antithrombin
- Degrades thrombin and factors IXa and Xa
- Activates tissue plasminogen activator (tPA)
- Clinical relevance: antithrombin III deficiency (e.g., due to liver failure or kidney failure)
- Nonspecific inhibitors: protease inhibitors in plasma (e.g., alpha-1-antitrypsin, alpha-2-macroglobulin)
- Drug-induced: anticoagulant treatment (see “Oral anticoagulants” and “Parenteral anticoagulants”)
-
Others
- Protein Z (factor X inhibitor)
- Heparin-like glycosaminoglycans (boosts antithrombin)
- Heparin cofactor II (requires heparin for activation)
Diseases that affect the inhibitors of the coagulation cascade may lead to hypercoagulability.

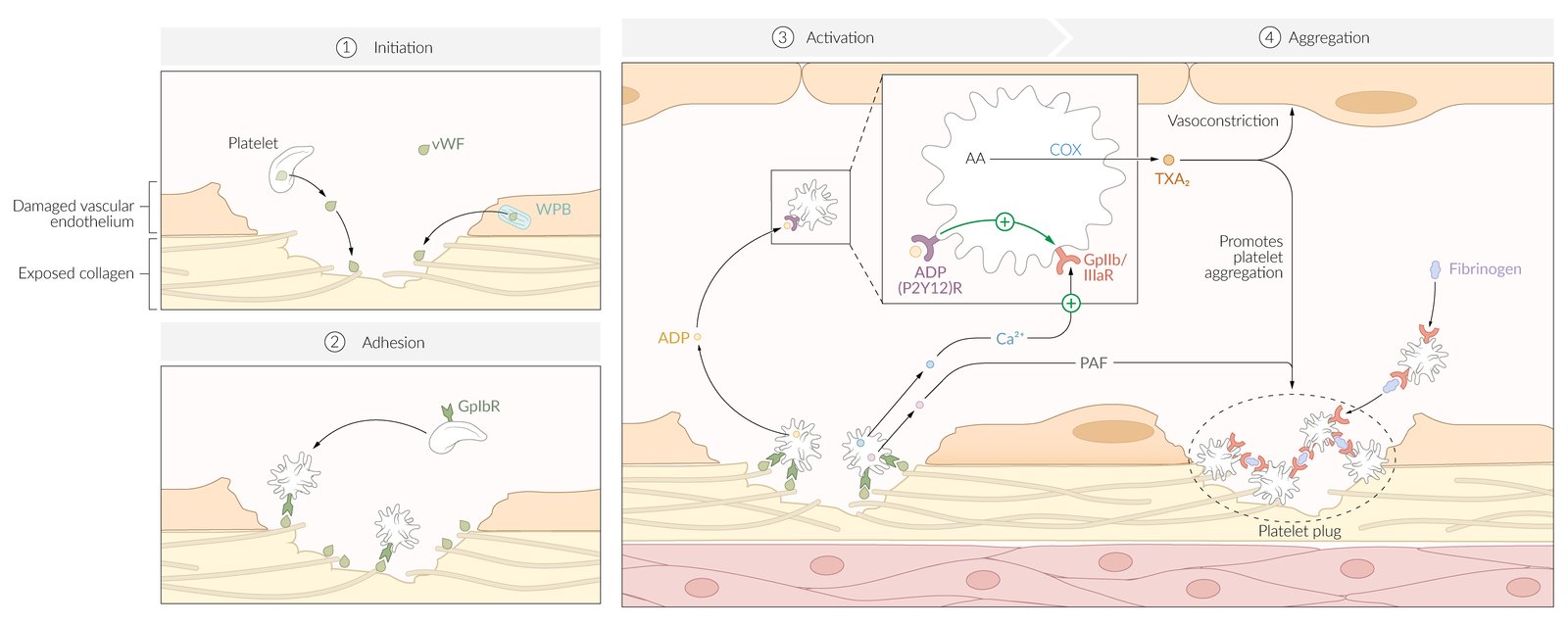
(1) Tissue injury results in the release of von Willebrand Factor (vWF) from Weibel Palade bodies (WPB) in endothelium and α-granules in platelets. VWF subsequently binds to exposed collagen at the site of injury. Vasoconstriction (not shown here) occurs through both a neural stimulation reflex and a release of endothelin from damaged endothelial cells.
(2) Platelets bind to exposed vWF at the site of injury and undergo a conformational change.
(3) Activated platelets release a combination of factors, including adenosine diphosphate (ADP), calcium ions (Ca2+), and platelet-activating factor (PAF). ADP binds to P2Y12 receptors on platelets, inducing a translocation of GpIIb/IIIa receptors to the cell surface. These receptors are promoted by Ca2+ ions.
(4) The expression of GpIIb/IIIa receptors allows activated platelets to crosslink with fibrinogen as an intermediary. The action of PAF along with thromboxane A2 (TXA2) further promotes platelet aggregation.
Platelet plugs are reinforced with fibrin during secondary hemostasis.
© AMBOSS
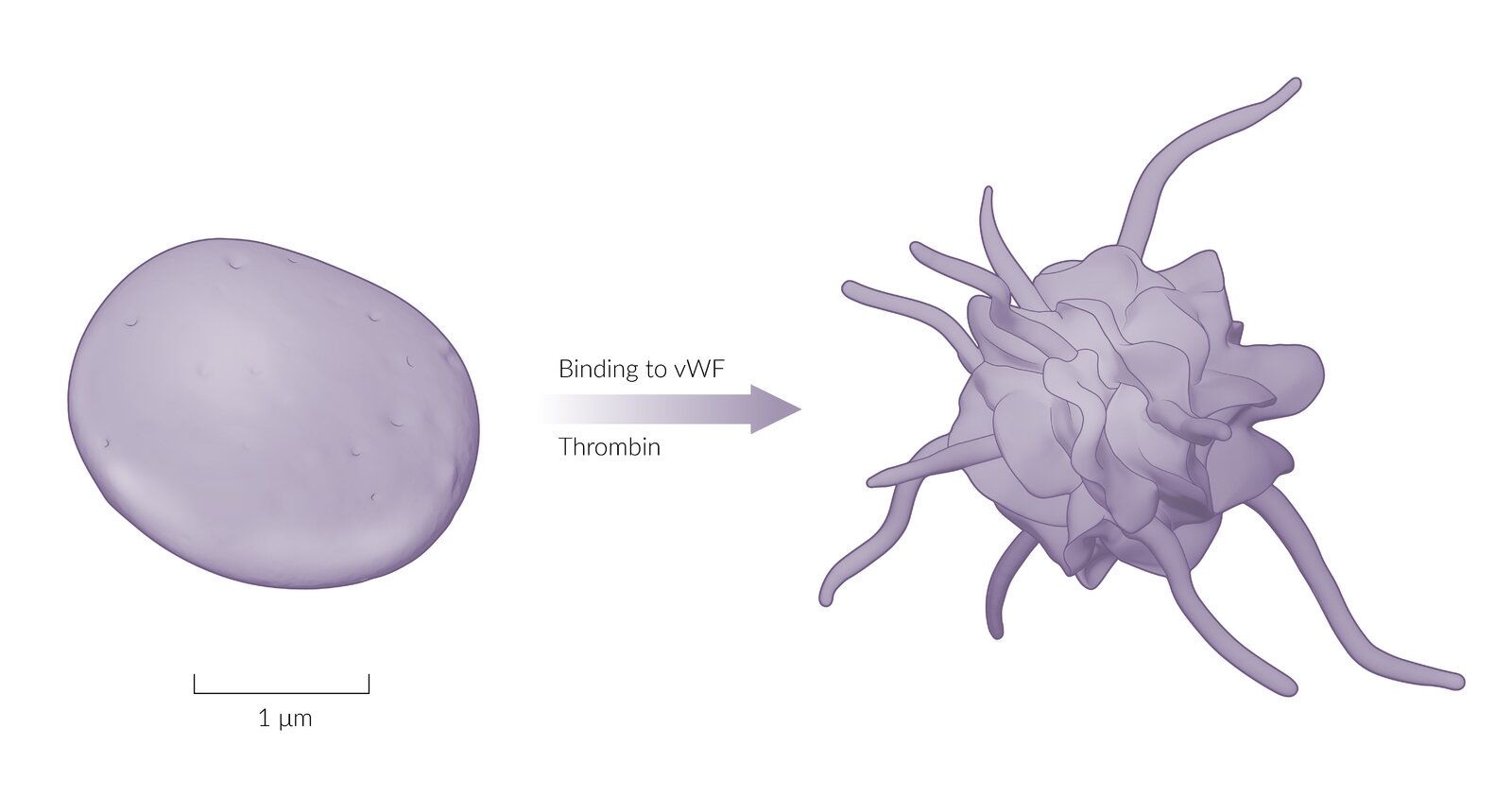
Sketches of platelets as seen on electron microscopy: Prior to activation, platelets have a discoid shape. When platelets become activated by von Willebrand factor or thrombin, they develop cytoplasmic, dendritic projections, which are called pseudopodia. They increase the platelet's surface area, which facilitates adhesion to endothelial lesions.
© AMBOSS

Link to questions: https://www.youtube.com/watch?v=vNq7oSnlwkc
© AMBOSS

© AMBOSS
© AMBOSS
© AMBOSS
© AMBOSS
Link to questions: https://www.amboss.com/us/collection/sample/secondary-hemostasis
© AMBOSS
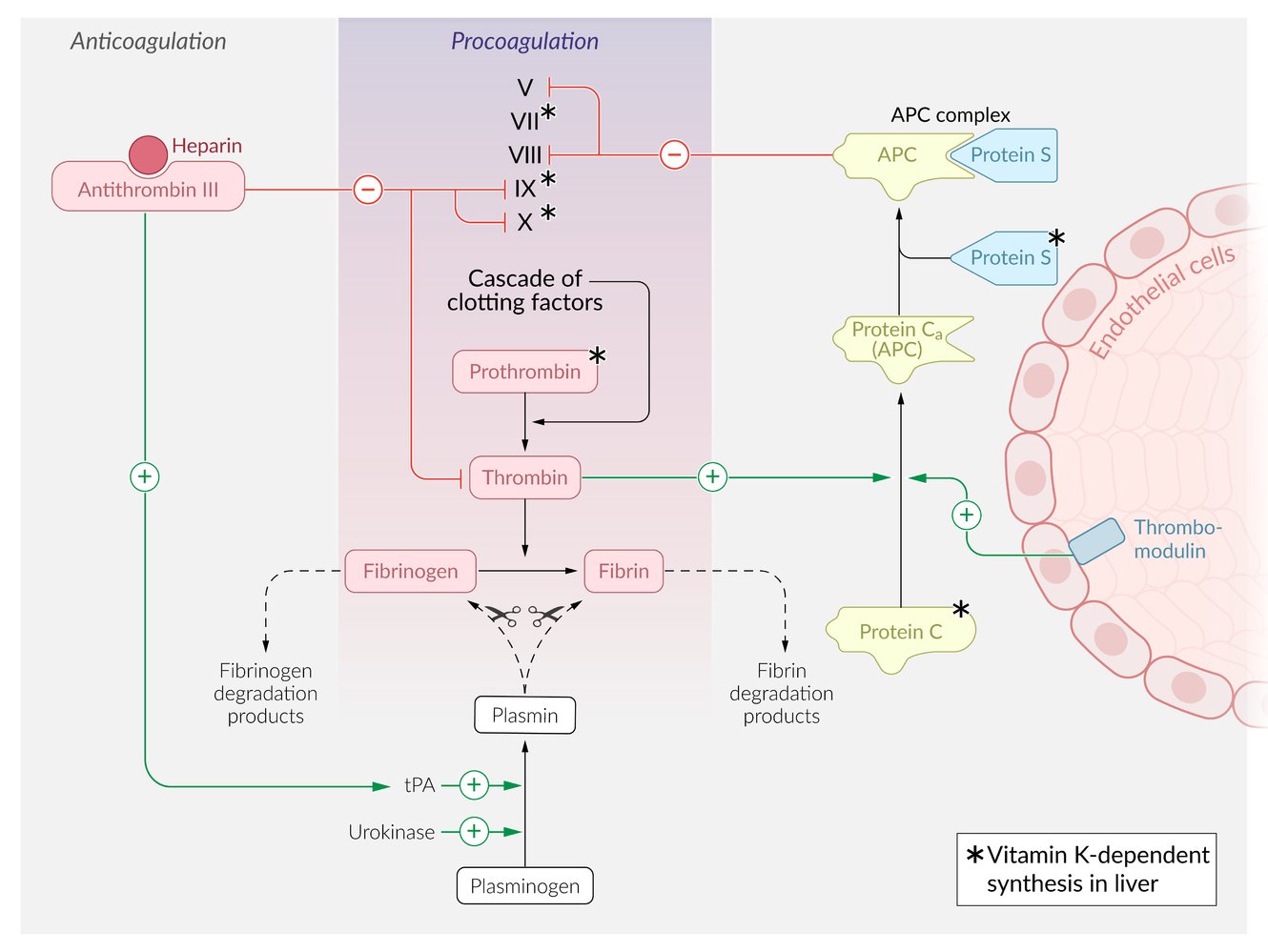
Clotting homeostasis is maintained through a balance of procoagulation (purple) and anticoagulation (gray) processes. The procoagulation pathway ultimately results in the enzymatic conversion of fibrinogen into fibrin, which is an integral part of blood clot formation. The procoagulation pathway is physiologically inhibited through two distinct anticoagulation pathways, using either antithrombin III or the APC complex.
Antithrombin III inactivates factors IX, X, and thrombin. This process is accelerated in the presence of heparin. Additionally, antithrombin III indirectly increases the activation of plasmin through the action of tissue plasminogen activator (tPA), resulting in the degradation of both fibrinogen and fibrin.
The APC complex pathway involves the activity of thrombomodulin, a membrane protein expressed on endothelial cells that activates protein C. Activated protein C then binds to protein S to form the APC complex, which inactivates procoagulation factors V and VIII. The procoagulation factor thrombin can also activate protein C, which results in a negative feedback loop on the procoagulation pathway.
© AMBOSS
Fibrinolysis
Overview [2]
- Definition: degradation of the fibrin network of thrombi by the enzyme plasmin
-
Mechanism
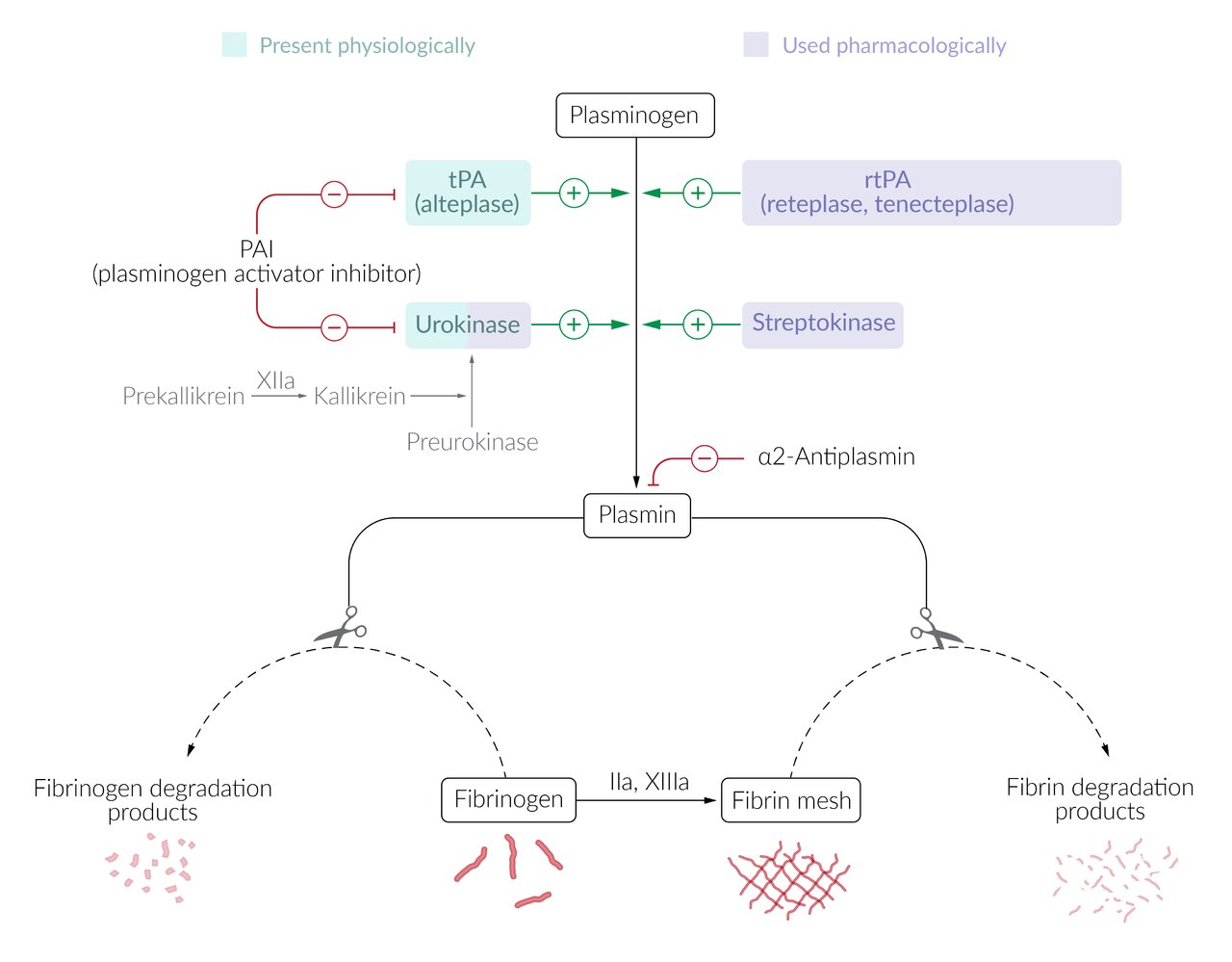
- Tissue injury leads to the release and activation of plasminogen activators, which convert plasminogen to its active form plasmin.
- Tissue plasminogen activator (tPA)
- Urokinase (see fibrinolytics)
- Plasmin breaks down and deactivates fibrin and fibrinogen → release of fibrin degradation products (e.g, D-dimers)
- Tissue injury leads to the release and activation of plasminogen activators, which convert plasminogen to its active form plasmin.
-
Regulation
- Plasminogen activator inhibitors (e.g., PAI-1) inhibit tPA
- Plasmin inhibitors (e.g., PPIC)
Fibrinolytic therapy [4]
-
Agents: Fibrinolytics promote the degradation of thrombi by activating plasminogen to plasmin.
-
Fibrin-specific agents
- Tissue plasminogen activator (tPA)
-
Recombinant plasminogen activators (rtPA): tPAs that are produced by recombinant biotechnology techniques
- Alteplase
- Reteplase (rPA)
- Tenecteplase (TNK-tPA)
-
Non-fibrin-specific agents
- Streptokinase: enzyme produced by group A streptococci; catalyzes the conversion of plasminogen to plasmin
- Urokinase: serine protease found in plasma, urine, and various types of tissue that is also used in fibrinolytic therapy
-
Fibrin-specific agents
- Mechanism of action: : directly or indirectly increase the concentration of plasmin; → cleavage of thrombin and fibrin [5]
-
Laboratory findings
- ↑ PT
- ↑ PTT
- Normal platelet count
-
Adverse effects
- Bleeding (e.g., intracranial hemorrhage)
- Hypersensitivity reactions (esp. to streptokinase) [6]
-
Indications
- STEMI (for details regarding fibrinolysis in STEMI, see “Fibrinolytic therapy in STEMI”)
- Ischemic stroke (for details regarding fibrinolysis in ischemic stroke, see “Reperfusion therapy for ischemic stroke”)
- Massive pulmonary embolism
-
Contraindications to fibrinolytic therapy
- Active bleeding
- Prior intracranial hemorrhage
- Recent surgery
- Severe hypertension
- Known bleeding diathesis
-
Reversal of adverse effects
-
Antifibrinolytics: group of drugs that impair fibrinolysis, typically by interfering with plasmin formation
- Tranexamic acid: a synthetic lysine analog and inhibitor of plasminogen with antifibrinolytic action
- Aminocaproic acid: a lysine derivative and inhibitor of plasminogen activators and plasmin with antifibrinolytic action
- Fresh frozen plasma (FFP), PCC, or cryoprecipitate (Cryoprecipitate is obtained from frozen blood plasma via centrifuge and contains more factor VIII and fibrinogen than FFP.)
- Platelet transfusions (if necessary)
-
Antifibrinolytics: group of drugs that impair fibrinolysis, typically by interfering with plasmin formation
Alteplase is a synthetic tissue plasminogen activator that converts plasminogen to plasmin. It is used in the treatment of STEMI, massive pulmonary embolism, and ischemic stroke.

Disorders of fibrinolysis
Hypoplasminogenemia [7]
- Definition: inherited genetic condition characterized by abnormally low levels of plasminogen, which results in a build-up of fibrin
- Epidemiology: rare disease; prevalence is estimated to be approx. 1:625,000
- Etiology: autosomal recessive inherited mutations in the PLG gene
- Pathophysiology: : mutations of the PLG gene → ↓ concentrations and/or functional impairment of plasminogen → ↓ plasmin → ↓ fibrinolysis → accumulation of fibrin
-
Clinical features: usually manifest in early infancy
- Formation of inflamed, thick, wood-like growths (ligneous pseudomembranes) on mucous membranes (especially the conjunctiva) that appear white, yellow, or red
- Other areas that can be affected: skin, CNS, gingiva, gastrointestinal tract, lungs, female genital tract (e.g., fallopian tubes)
- Not associated with thrombophilia
-
Diagnostics
- Initially: clinical presentation, laboratory tests for plasminogen activity
- Genetic testing confirms the diagnosis.
- Differential diagnosis: infections (most common), but also conditions such as allergies, GERD, IBD, endometriosis, depending on the area affected
-
Treatment
-
Intravenous injection of ryplazim [8]
- Plasma-derived human plasminogen used in the treatment of hypoplasminogenemia
- Adverse effects include gastrointestinal upset, extremity pain, and hemorrhage.
- Surgical removal of growths is usually not recommended due to rapid regrowth.
-
Intravenous injection of ryplazim [8]
-
Complications
- Corneal scarring, loss of vision
- Duodenal ulcers
- Occlusive hydrocephalus
- Can reduce fertility
- Impaired wound healing
© AMBOSS
Etiology
Hemorrhagic diathesis is the abnormally increased susceptibility to bleeding.

Disorders of primary hemostasis
-
Platelet disorders [9]
- Platelet deficiency (see “Etiology” in “Thrombocytopenia”)
-
Platelet dysfunction (thrombocytopathy): disorders that lead to dysfunctional adhesion or aggregation of platelets
-
Inherited
- Von Willebrand disease
- Bernard-Soulier syndrome
- Glanzmann thrombasthenia
-
Acquired
- Drug-induced: e.g., aspirin, NSAID, clopidogrel
- Immune thrombocytopenic purpura
- Chronic kidney disease
- Cardiopulmonary bypass [10]
-
Inherited
-
Disorders affecting the vessel wall
- Vascular hemorrhagic diathesis (e.g., IgA vasculitis, hereditary hemorrhagic telangiectasia)
- Thrombotic microangiopathy (e.g., HUS and TTP)
- Conditions with impaired collagen synthesis (e.g., scurvy, Ehlers-Danlos syndrome)
| Differential diagnosis of platelet disorders | ||||||
|---|---|---|---|---|---|---|
| HUS | TTP | Disseminated intravascular coagulation (DIC) | Immune thrombocytopenic purpura (ITP) | Bernard-Soulier syndrome | Glanzmann thrombasthenia | |
| Pathophysiology |
|
|
|
|
|
|
| Typical presentation |
|
|
|
|
||
| Peripheral smear |
|
|
|
|
|
|
| PT (INR) and aPTT |
|
|
|
|||
| D-dimer, fibrin degradation products |
|
|
|
|||

Disorders of secondary hemostasis (disorders of the coagulation cascade)
-
Intrinsic pathway
- Factor VIII deficiency (hemophilia A)
- Factor IX deficiency (hemophilia B)
- Factor XI deficiency (hemophilia C)
- Extrinsic pathway: : factor VII deficiency (autosomal recessive bleeding disorder caused by mutation of the F7 gene)
-
Both pathways
-
Deficiency or inhibition of vitamin K-dependent coagulation factors II, VII, IX, and X
- Vitamin K deficiency: e.g., malabsorption syndrome, depletion of gut flora (e.g., following antibiotic administration), vitamin K deficiency bleeding of the newborn
- Vitamin K antagonist therapy (e.g., warfarin)
- Inhibition of coagulation factors by autoantibodies (most commonly anti-factor VIII) [11]
- Disseminated intravascular coagulation (DIC)
- Impaired hepatic production of coagulation factors (e.g., cirrhosis)
- Fibrinogen deficiency
- Anticoagulant treatment
-
Deficiency or inhibition of vitamin K-dependent coagulation factors II, VII, IX, and X

In disorders of primary hemostasis, platelet aggregation is impaired, whereas in disorders of secondary hemostasis it is the coagulation cascade that is impaired.
Hyperfibrinolysis [12]
- Definition: excessive fibrinolytic activity, resulting in increased bleeding
-
Etiology
- Disseminated intravascular coagulation (secondary to liver dysfunction, sepsis, etc.)
- Peripartum complications
- Treatment with fibrinolytics
- Surgery in tPA-rich organs such as the prostate and uterus
- Prostate carcinoma (paraneoplastic)
- Pathophysiology: excessive plasmin activity → increased fibrin degradation → thrombus instability and dissolution shortly after formation
- Treatment: antifibrinolytic agents (e.g., tranexamic acid) if DIC has been excluded
Hypofibrinolysis
- Definition: abnormally low fibrinolytic activity, resulting in thrombosis
-
Etiology
- tPA or uPA deficiency
- Overexpression of PAI-1 or TAFI
Hypofibrinogenemia [13]
- Definition: : a disorder characterized by low plasma fibrinogen levels
-
Etiology
- Congenital: mutation of fibrinogen genes (FGA, FGB, FGG) on chromosome 4
- Acquired: associated with liver failure, disseminated intravascular coagulation (DIC), liver transplantation, massive blood transfusion, hemodilution, consumption of clotting factors [14]
-
Pathophysiology
- Congenital: mutation → lack of functional fibrinogen glycoprotein → ↓ production of fibrinogen → hemorrhagic diathesis
- Acquired: ↓ fibrinogen concentration (various causes) → hemorrhagic diathesis
- Symptoms and findings: epistaxis, gastrointestinal hemorrhage, gingival bleeding
- Treatment: fresh frozen plasma

© AMBOSS
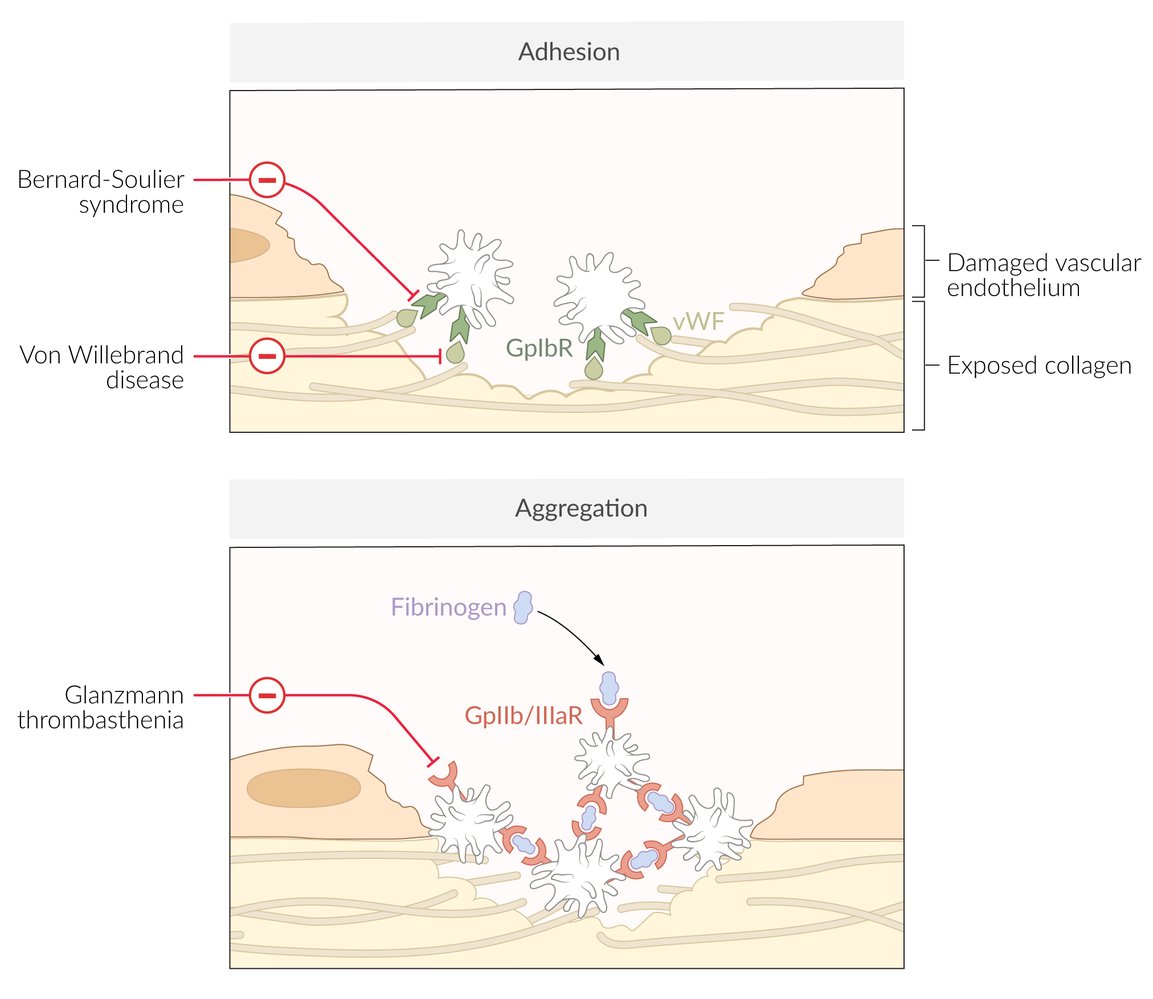
Red indicators link syndromes with their associated defective or absent molecules, according to which phase of primary hemostasis is affected (i.e., adhesion or aggregation)
© AMBOSS
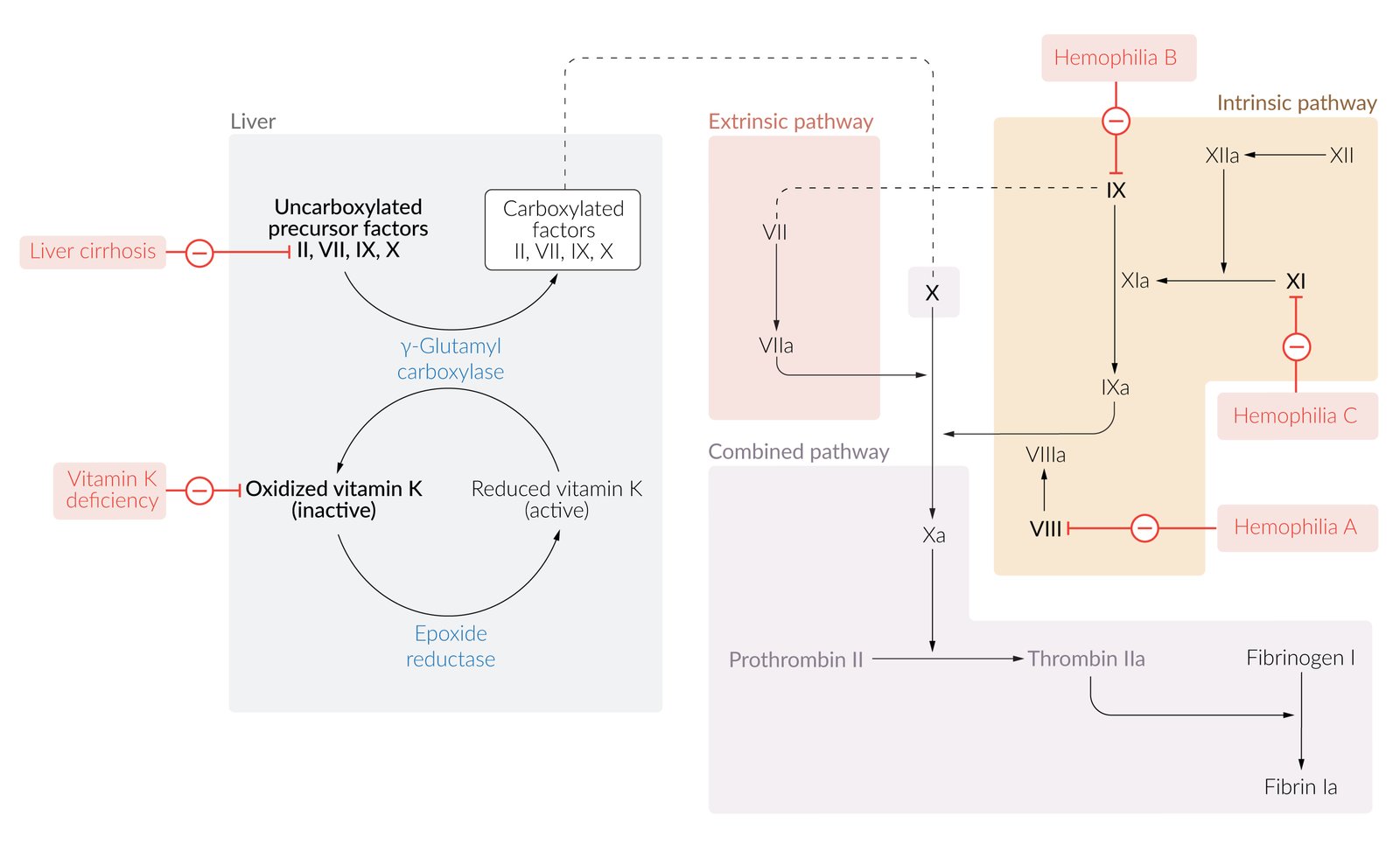
© AMBOSS
Clinical features
| Clinical features of bleeding disorders [15][16] | ||
|---|---|---|
| Disorder | Characteristics of bleeding | Manifestations |
| Primary hemostasis disorders [9] |
|
|
| Secondary hemostasis disorders |
|
|
| Disseminated intravascular coagulation |
|
|
- A thorough physical examination is essential for diagnosing bleeding disorders and should include the inspection of the entire skin, mucosa (esp. oral cavity), and joints.
- Watch out for signs of physical abuse, which may produce patterns of bruising that resemble those of bleeding disorders. Signs of physical abuse include:
- Inconsistency of clinical findings and history
- Atypical bruising patterns (e.g., on back, forehead, ears, neck) and/or retinal hemorrhages in children ≤ 4 years (see “Physical child abuse”)
Superficial, petechial bleeding indicates defects of primary hemostasis, whereas large, palpable ecchymoses and deep tissue bleeding suggest defects of secondary hemostasis!


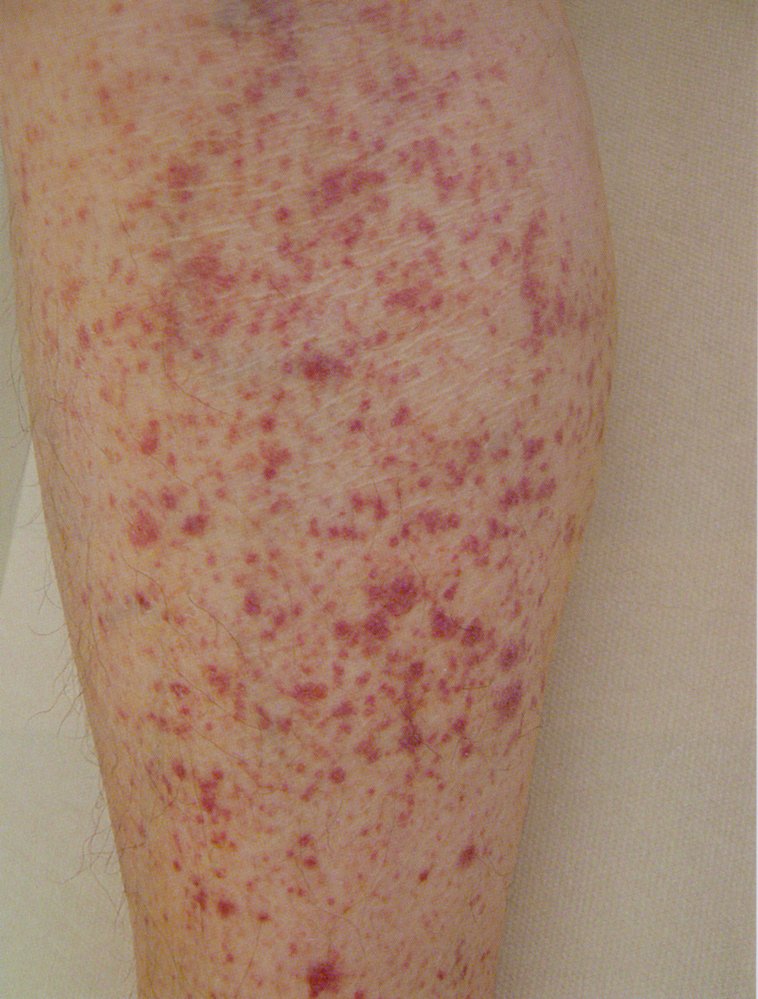
The lower leg displays numerous red spots, each about the size of a pinhead, that do not blanch or disappear when pressed. These are indicative of petechial hemorrhages related to thrombocytopenia.
Source: © IMPP
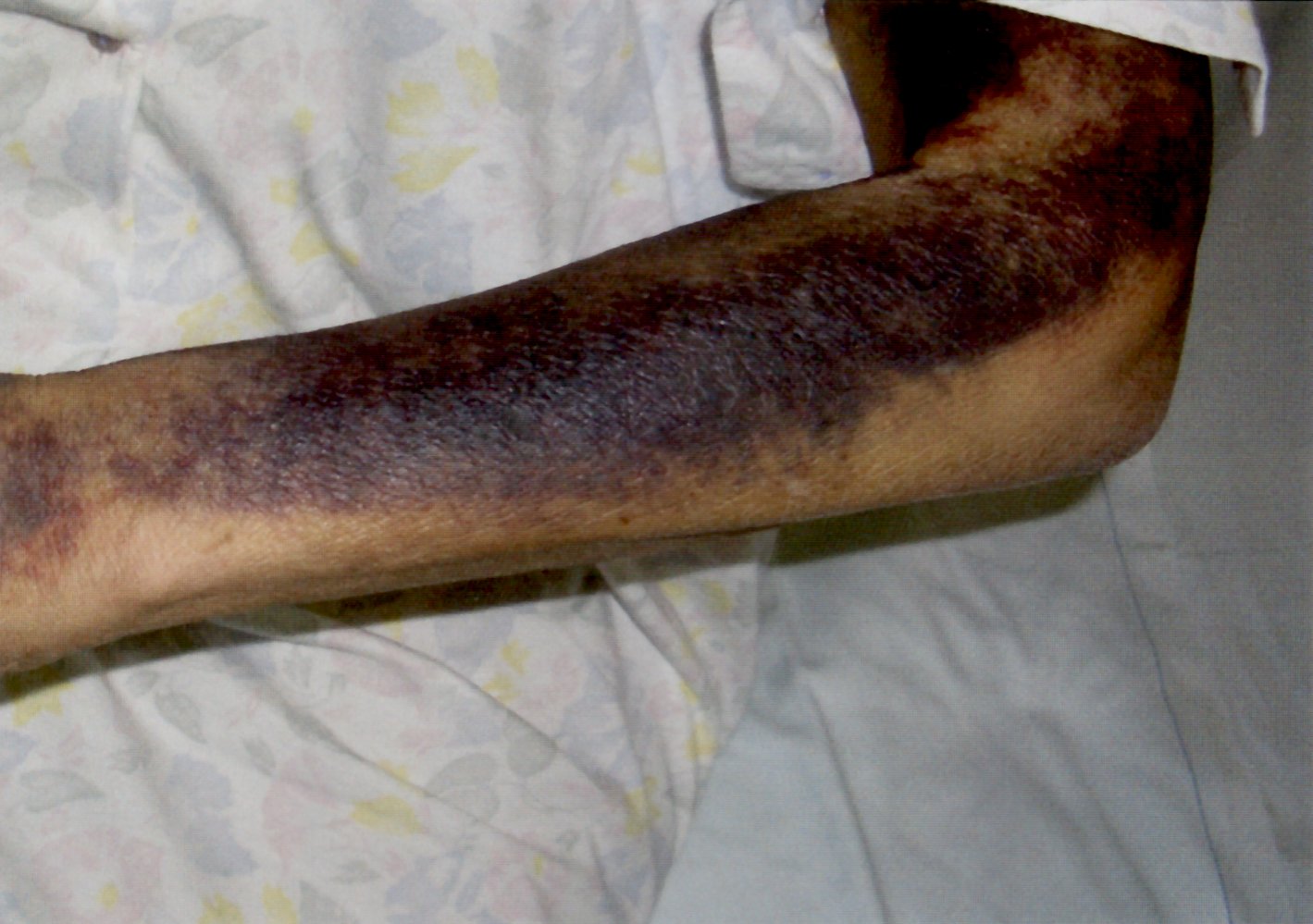
Diffuse ecchymoses are visible on the radial aspect of the forearm.
The hemorrhage is extensive and diffuse, which indicates an impairment of secondary hemostasis.
Specific antibodies that reduce factor VIII activity inhibit secondary hemostasis in individuals with systemic lupus erythematosus.
Source: © IMPP
Diagnosis
Clinical assessment and basic laboratory testing are used to differentiate between primary and secondary disorders of hemostasis. Advanced testing is used to identify the specific disorder.
Clinical assessment [17][18][19]
-
Symptoms
- Ask about features of mucosal, skin, and deep-tissue bleeding (see “Clinical features”).
- Quantify bleeding severity with a bleeding assessment tool.
-
Features associated with bleeding disorders
- Known bleeding disorder in a family member
- Chronic illness: e.g., chronic kidney disease, liver disease, malignancy, autoimmune diseases, malabsorption
- Acute illness: e.g., recent infections , sepsis , use of mechanical circulatory support
- Poor nutritional status
- Pregnancy
-
Use of medications
- Antithrombotic agents: anticoagulants, antiplatelet agents
- Other drugs that can affect platelet function and/or count, including:
- Chemotherapeutic agents
- NSAIDs
- Phosphodiesterase inhibitors (e.g., cilostazol, sildenafil, theophylline)
- Beta-lactam antibiotics
- Antimycotics and antibiotics (e.g., miconazole, nitrofurantoin)
- Nitrates
- Antidepressants
Most bleeding disorders are acquired; i.e., secondary to medications or associated with acute or chronic illness. [17]
Women presenting with menorrhagia should also be evaluated by a gynecologist.
Bleeding assessment tools (BATs) [20][21][22]
BATs are questionnaires that standardize the assessment of bleeding symptoms and identify patients who may benefit from advanced testing. Multiple tools are available.
- The International Society of Thrombosis and Hemostasis BAT includes the following symptoms: [21][22][23]
- Mucocutaneous symptoms: cutaneous signs of bleeding, bleeding from minor wounds, epistaxis, bleeding in the oral cavity
- Bleeding after procedures: after a surgery or dental extraction
- Musculoskeletal bleeding: hemarthroses, or muscular hematomas
- CNS bleeding
- GI bleeding
- Hematuria
- Gynecological bleeding: menorrhagia or postpartum hemorrhage
- Each symptom receives a score based on its severity.
- An elevated total score should prompt evaluation for bleeding disorders. [22][24] [25][26]
- A low score does not exclude bleeding disorder.
Initial laboratory studies [18][19][20][27]
-
Indications
- History or physical examination suggestive of a bleeding disorder
- Elevated BAT score
-
Studies
- CBC, BMP, liver chemistries
-
Peripheral blood smear
- Platelet clumping: suggests pseudothrombocytopenia
- Schistocytes or other fragmented cells: consistent with microangiopathic hemolytic anemia (e.g., thrombotic thrombocytopenic purpura)
- Identification of blasts: suggests hematologic malignancies
- Coagulation panel; including fibrinogen level: Elevated PT/INR and/or aPTT are suggestive of disorders of secondary hemostasis.
- Platelet function analysis (PFA): Abnormal platelet function (or, if not available, a prolonged bleeding time; ) suggests a disorder of primary hemostasis. [19]
- vWF antigen and vWF activity (vWF ristocetin activity): for diagnosis of vWD
Von Willebrand disease is the most common inherited bleeding disorder, affecting up to 1% of the population. Von Willebrand factor concentration and vWF activity are now commonly part of initial diagnostic studies. [20][27]
| Interpretation of laboratory findings in bleeding disorders [18][20][28] | |||||
|---|---|---|---|---|---|
| Defective pathway | Disorders | Platelet count | PFA or bleeding time | PT/INR | aPTT |
| Disorders of primary hemostasis | Thrombocytopenia |
|
|
|
|
| Platelet dysfunction (e.g., aspirin therapy, chronic kidney disease) |
|
|
|
|
|
| Disorders of secondary hemostasis | Extrinsic pathway (e.g., factor VII deficiency) |
|
|
|
|
| Intrinsic pathway (e.g., hemophilia, heparin therapy) |
|
|
|
|
|
| Intrinsic and extrinsic pathways (e.g., deficiency of vitamin K-dependent coagulation factors) |
|
|
|
|
|
| Disorders of primary AND secondary hemostasis | Von Willebrand disease |
|
|
|
|
| Disseminated intravascular coagulation (DIC) |
|
|
|
|
|
DIC is characterized by thrombosis, hemorrhage, and organ dysfunction and can be a medical emergency that requires immediate treatment.
A detailed clinical assessment and initial laboratory studies are sufficient to diagnose the most common disorders of hemostasis (e.g., platelet dysfunction secondary to medications or bleeding disorders associated with acute or chronic disease).
Advanced laboratory studies [18][19]
Advanced laboratory studies help identify specific disorders. The choice of studies depends on the suspected underlying pathology and usually requires specialist consult.
- Disorders of primary hemostasis: low platelet count or abnormal platelet function test with normal INR and PTT
- Disorders of secondary hemostasis: abnormal INR or PTT with normal platelet counts and platelet function
- Unclear after initial studies: normal platelet count and function and normal coagulation panel but high clinical suspicion for a bleeding disorder
Suspected disorders of primary hemostasis
-
Low platelet count
- Base further testing on clinical suspicion. For example:
- Thrombocytopenia in a pregnant patient: Start investigations for HELLP syndrome.
- Anemia and thrombocytopenia in a patient with suggestive symptoms: Start investigations for TTP.
- Presence of B symptoms or other abnormalities on CBC and/or blood smear: Start investigations for a hematologic malignancy.
- See “Diagnostics” in “Thrombocytopenia” for details.
- Base further testing on clinical suspicion. For example:
-
Abnormal platelet function [29]
- Acquired (most common): etiology (e.g., medications or chronic illness) usually clear after initial evaluation
- Inherited (rare): advanced testing to further assess platelet function and identify the etiology
- See also “Differential diagnoses of platelet disorders.”
Suspected disorders of secondary hemostasis [19][30]
- Congenital factor deficiencies: confirmed with factor assays (low factor concentrations)
- Presence of factor inhibitors: can be diagnosed using mixing studies
| Possible mechanisms and coagulation factors affected in disorders of secondary hemostasis [19][30] | ||
|---|---|---|
| Elevated PT/INR | Normal PT/INR | |
| Elevated aPTT |
|
|
| Normal aPTT |
|
|
Disorder unclear after initial testing [19]
Additional testing is required if suspicion of a bleeding disorder is high but coagulation testing is normal. Possible etiologies include:
- Hyperfibrinolysis: Testing includes alpha 2-antiplasmin level and plasminogen activation inhibitor-1 level. [31]
- Vascular disorders (e.g., Ehler Danlos syndrome, hereditary hemorrhagic telangiectasia): Genetic testing is required.
- Factor XIII deficiency
- Combined coagulation factor deficiency
If the diagnosis remains unclear, repeat testing for vWD is recommended. [19]
Treatment
Treatment should be guided by a specialist and target the specific cause of the bleeding. Occasionally, the transfusion of blood products may be indicated. For patients with acute or major bleeding, see “Management of acute bleeding” below.
General principles [30][32]
- Avoid medications that may impair hemostasis.
- Ensure optimal preventive medical and dental health care.
- Consult a specialist to determine any preventive measures that are necessary before invasive procedures.
- Provide patient education on lifestyle modification in the setting of bleeding disorders.
- Consider screening for HIV and hepatitis if the patient has a history of multiple transfusions.
Disorders of primary hemostasis [33][34]
Platelet transfusion
- Used in the treatment of both thrombocytopenia and platelet dysfunction
- Indications
- Active bleeding
- Prevention of bleeding during invasive procedures
- Prophylactically in patients with extremely low platelet count [30]
- Contraindications: uremic platelet dysfuntion
Disease-specific treatment
-
Uremic platelet dysfunction [35]
- Preventative measures include dialysis, erythropoietin, and conjugated estrogens. [36]
- Desmopressin: may be used prior to invasive procedures or to treat active bleeding
- Platelet transfusion is not indicated due to rapid inactivation of transfused platelets
- Drug-related platelet dysfunction [33]
- Discontinue medications causing dysfunction.
- Consider desmopressin
- Immune thrombocytopenia: Treatment may include corticosteroids and IVIG. Platelet transfusion is typically ineffectual.
- Heparin-induced thrombocytopenia: Stop all heparin and initiate an alternative form of anticoagulation.
- HELLP syndrome: Consult obstetrician immediately for specific management.
One random donor platelet unit typically increases platelet count by 5,000–10,000/mm3 in a 70-kg patient. One apheresis unit (the equivalent of 6–8 single units) increases platelet count by 30,000–40,000/mm3. [37]
Disorders of secondary hemostasis
Inherited coagulation factor deficiency [27]
- Depending on the etiology, treatment can include one or more of the following:
- Blood product transfusions
-
Replacement of specific coagulation factors, e.g.:
- Factors VIII, IX, or XI (e.g., for hemophilia)
- Concentrates containing vWF and factor VIII (for vWD)
- Pharmacological therapy (e.g., desmopressin, antifibrinolytics)
- Monoclonal antibody therapy
- See "Management of hemophilia" and "Treatment of von Willebrand disease" for specific recommendations.
Acquired coagulation factor deficiency
-
Vitamin K deficiency [38]
- Dietary deficiency
- Vitamin K replacement: phytonadione
- Optimize enteral or parenteral nutrition.
- Associated with anticoagulant use: See “Anticoagulant reversal” for specific recommendations.
- Dietary deficiency
-
Liver disease: complex management [39][40][41]
- Consult gastroenterology and hematology early.
- Consider vitamin K replacement for patients with abnormal coagulation studies. [40][42]
- Screen for and manage comorbid conditions that affect hemostasis (e.g., uremia, bacterial overgrowth, portal hypertension, thrombocytopenia).
The effect of liver disease on hemostasis is complex: The clinical effects of thrombocytopenia and reduced coagulation factor synthesis are often negated by a simultaneous decrease in the production of profibrinolytic factors. Bleeding often does not occur, even if the laboratory values of hemostasis are markedly abnormal. [39]
Impaired coagulation factor function
- Therapeutic or accidental anticoagulant use: See “Anticoagulant reversal.”
- Coagulation factor inhibitors: Treatment includes immunosuppression (e.g., glucocorticoids) and occasional use of bypassing agents. [43]
Patients with an inhibitor to a coagulation factor are at high risk of severe or fatal bleeding. [43]
Management of acute bleeding
Management [17]
- Start ABCDE approach and stabilize the patient as needed.
- Establish appropriate IV access.
- Consult hematology service.
- Assess the need for anticoagulant reversal, discontinue ongoing anticoagulants and platelet inhibitors, unless contraindicated.
- Consider consultation for a procedural intervention to control bleeding (e.g., upper endoscopy, colonoscopy, surgery).
-
Start transfusion of blood products if needed (see also “Massive transfusion”).
- pRBCs: Target Hb using a liberal or restrictive transfusion strategy.
- Platelet transfusion: for thrombocytopenia or as part of a massive transfusion protocol
- Fresh frozen plasma (FFP), cryoprecipitate : as part of massive transfusion protocol or to replace coagulation factors and/or fibrinogen
- Prothrombin complex concentrate: Use only with expert guidance; potentially fatal thrombotic complications may occur. [44][45]
-
Consider replacement of specific coagulation factors
- Known hemophilia: factors VIII, IX, or XI
- Known von Willebrand disease (vWD): concentrates containing vWF and factor VIII
-
Consider adjuvant drug therapy depending on suspected etiology
- Anticoagulant reversal, e.g., Vitamin K (phytonadione ) [38]
- Desmopressin [46]
- Antifibrinolytics (e.g., for hyperfibrinolysis)
Diagnostics should not delay factor replacement therapy in patients with known bleeding disorders presenting with major bleeding. [47][48][49]
Patients with a suspected disorder of secondary hemostasis presenting with major bleeding can benefit from the transfusion of fresh frozen plasma while diagnostic studies are performed. [17]
Diagnostics [17]
-
Routine testing
- CBC with blood smear, BMP, liver chemistries, and type and cross
- Coagulation studies: Obtain a coagulation panel and measure vWF and vWF activity.
-
Viscoelastic hemostatic assays (VHAs): a point-of-care test with rapid results that provides a comprehensive assessment of hemostatic function [50][51][52]
- Common modalities: thromboelastogram (TEG®), rotational thromboelastometry (ROTEM®), and Sonoclot®
- Uses [52][53][54][55][56]
- Rapid bleeding (e.g., cardiac and liver transplant surgery, trauma, DIC, peripartum hemorrhage)
- Complex hemostasis disorders (e.g., liver disease)
")
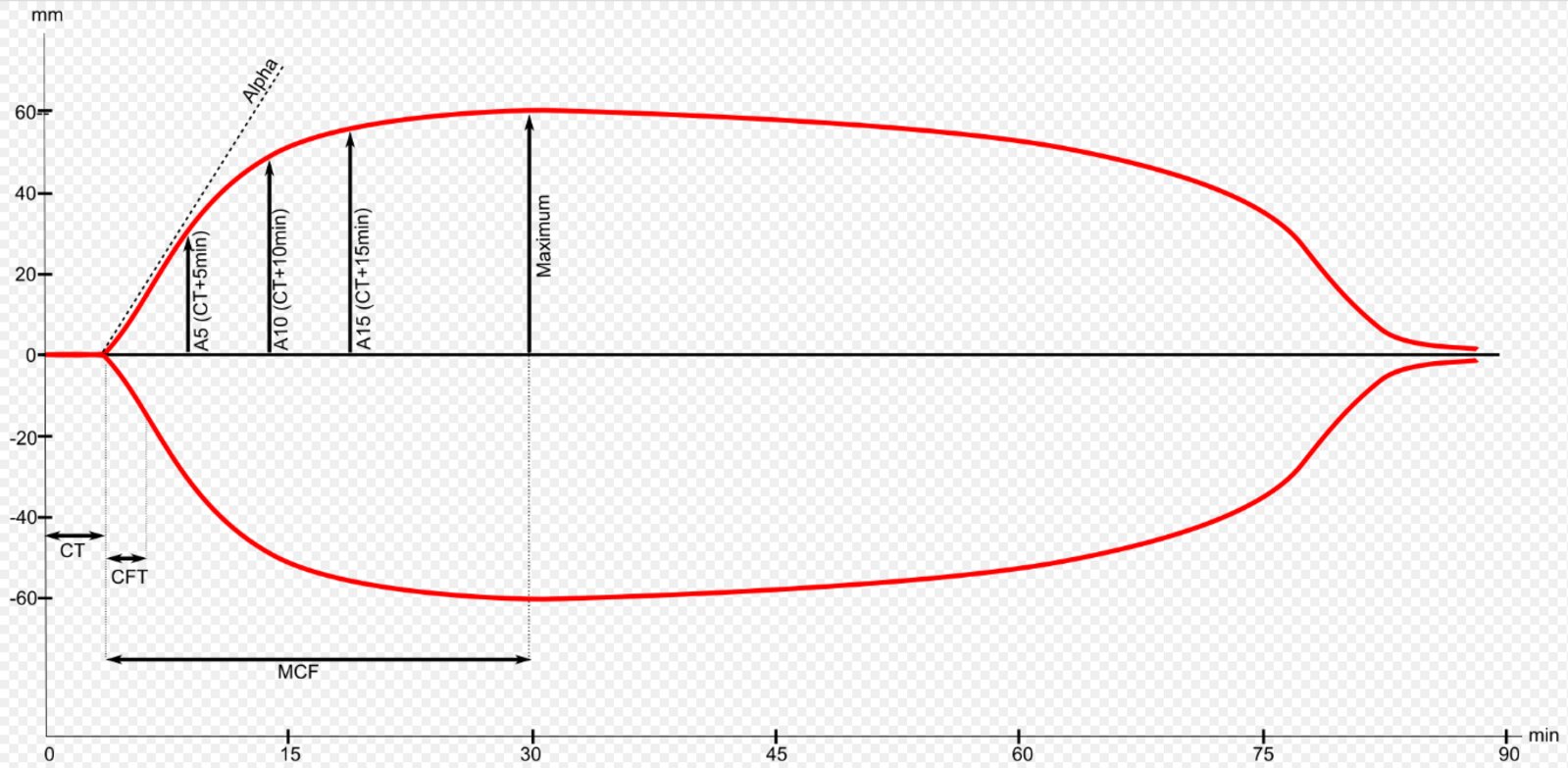
Normal thromboelastogram with parameters
For thromboelastography, activators are added to a blood sample in order to induce and measure coagulation. Then firmness and elasticity of the clot are measured over time. Clotting-time (CT) is the time until initial clot formation. Clot-formation-time (CFT) is the time from initial clot formation until reaching 20 mm in strength. The alpha angle is a measure of the pace of clot strengthening. The maximum clot strength is described by the maximum clot-firmness (MCF).
The parameters are used to examine blood coagulation.
Source: “Thrombelastometrie ROTEM” by A1b2 commons, Wikimedia Commons, licensed under CC BY-SA 4.0.
{kind=link}
External Resources
References
- "Overview of Platelet Disorders". http://www.merckmanuals.com/professional/hematology-and-oncology/thrombocytopenia-and-platelet-dysfunction/overview-of-platelet-disorders. [2014-09-01]
- Weerasinghe A, Taylor KM. "The platelet in cardiopulmonary bypass". Ann Thorac Surg. 66(6). :2145-2152. (1998)
- Franchini M, Castaman G, Coppola A, et al. "Acquired inhibitors of clotting factors: AICE recommendations for diagnosis and management". Blood Transfus. 13(3). :498-513. (2015)
- Fay WP, Leung LLK, Tirnauer JS. "Thrombotic and Hemorrhagic Disorders due to Abnormal Fibrinolysis". UpToDate. UpToDate. https://www.uptodate.com/contents/thrombotic-and-hemorrhagic-disorders-due-to-abnormal-fibrinolysis. [2017-05-26]
- Thompson RJ, Portmann BC, Roberts EA. "Genetic and metabolic liver disease". Elsevier. :157-259. (2011). ISBN: 9780702033988
- Shaz BH, MD BH, Hillyer CD, MD CD, Gil MR. "Transfusion Medicine and Hemostasis". Elsevier Science. :1050. (2019). ISBN: 9780128137260
- Girolami A, Luzzatto G, Varvarikis C, et al. "Main clinical manifestations of a bleeding diathesis: an often disregarded aspect of medical and surgical history taking". Haemophilia. 11(3). :193-202. (2005)
- Drews RE. "Approach to the adult patient with a bleeding diathesis". UpToDate. UpToDate. https://www.uptodate.com/contents/approach-to-the-adult-patient-with-a-bleeding-diathesis?source=machineLearning&search=hemostasis%20disorders&selectedTitle=1~150§ionRank=2&anchor=H10#H6. [2017-01-11]
- Walls R, Hockberger R, Gausche-Hill M. "Rosen's Emergency Medicine". Elsevier Health Sciences. (2018). ISBN: 9780323354790
- Neutze D, Roque J. "Clinical Evaluation of Bleeding and Bruising in Primary Care". Am Fam Physician. 93(4). :279-86. (2016)
- Boender J, Kruip MJHA, Leebeek FWG. "A diagnostic approach to mild bleeding disorders". J of Thromb and Haemost. 14(8). :1507-1516. (2016)
- Rodeghiero F, Pabinger I, Ragni M, et al. "Fundamentals for a Systematic Approach to Mild and Moderate Inherited Bleeding Disorders: An EHA Consensus Report". Hemasphere. 3(5). (2019)
- RODEGHIERO F, TOSETTO A, ABSHIRE T, et al. "ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders". J of Thromb and Haemost. 8(9). :2063-2065. (2010)
- Adler M, Kaufmann J, Alberio L, Nagler M. "Diagnostic utility of the ISTH bleeding assessment tool in patients with suspected platelet function disorders". J Thromb Haemost. 17(7). :1104-1112. (2019)
- Elbatarny M, Mollah S, Grabell J, et al. "Normal range of bleeding scores for the ISTH-BAT: adult and pediatric data from the merging project". Haemophilia. 20(6). :831-835. (2014)
- Moenen FCJI, Nelemans PJ, Schols SEM, et al. "The diagnostic accuracy of bleeding assessment tools for the identification of patients with mild bleeding disorders: A systematic review". Haemophilia. 24(4). :525-535. (2018)
- Fasulo MR, Biguzzi E, Abbattista M, et al. "The ISTH Bleeding Assessment Tool and the risk of future bleeding". Journal of Thrombosis and Haemostasis. 16(1). :125-130. (2017)
- Lowe GC, Lordkipanidzé M, Watson SP. "Utility of the ISTH bleeding assessment tool in predicting platelet defects in participants with suspected inherited platelet function disorders". Journal of Thrombosis and Haemostasis. 11(9). :1663-1668. (2013)
- James PD, Goodeve AC. "von Willebrand disease". Genet Med. 13(5). :365-376. (2011)
- Bashawri LA, Ahmed MA. "The approach to a patient with a bleeding disorder: for the primary care physician.". Journal of family & community medicine. 14(2). :53-8. (2007)
- Paniccia R, Priora R, Alessandrello Liotta A, Abbate R. "Platelet function tests: a comparative review". Vasc Health Risk Manag. :133. (2015)
- Kruse-Jarres R, Singleton TC, Leissinger CA. "Identification and basic management of bleeding disorders in adults.". J Am Board Fam Med. 27(4). :549-64. (2014)
- Longstaff C. "Measuring fibrinolysis: from research to routine diagnostic assays". J Thromb Haem. 16(4). :652-662. (2018)
- Rydz N, James P. "Approach to the Diagnosis and Management of Common Bleeding Disorders". Semin Thromb Hemost. 38(07). :711-719. (2012)
- Konkle BA. "Acquired Disorders of Platelet Function". Hematology Am Soc Hematol Educ Program. 2011(1). :391-396. (2011)
- DiMichele DM, Hathaway WE. "Use of DDAVP in inherited and acquired platelet dysfunction.". Am J Hematol. 33(1). :39-45. (1990)
- Hedges SJ, Dehoney SB, Hooper JS, Amanzadeh J, Busti AJ. "Evidence-based treatment recommendations for uremic bleeding". Nat Clin Pract Nephrol. 3(3). :138-153. (2007)
- Fraser IS, Porte RJ, Kouides PA, Lukes AS. "A benefit-risk review of systemic haemostatic agents: part 1: in major surgery.". Drug Saf. 31(3). :217-30. (2008)
- Mohanty D. "Current concepts in platelet transfusion". Asian Journal of Transfusion Science. 3(1). :18. (2009)
- Schulman S, Furie B. "How I treat poisoning with vitamin K antagonists". Blood. 125(3). :438-442. (2015)
- Caldwell SH, Hoffman M, Lisman T, et al. "Coagulation disorders and hemostasis in liver disease: Pathophysiology and critical assessment of current management". Hepatology. 44(4). :1039-1046. (2006)
- Hunt BJ. "Bleeding and Coagulopathies in Critical Care". N Engl J Med. 370(9). :847-859. (2014)
- Lisman T, Porte RJ. "Pathogenesis, prevention, and management of bleeding and thrombosis in patients with liver diseases". Research and Practice in Thrombosis and Haemostasis. 1(2). :150-161. (2017)
- Kujovich JL. "Coagulopathy in liver disease: a balancing act". Hematology. 2015(1). :243-249. (2015)
- W Collins P, Chalmers E, et al. "Diagnosis and management of acquired coagulation inhibitors: a guideline from UKHCDO". Br J Haematol. 162(6). :758-773. (2013)
- Leung LLK, Mannucci PM, Tirnauer JS. "Overview of Hemostasis". UpToDate. UpToDate. https://www.uptodate.com/contents/overview-of-hemostasis. [2016-12-15]
- Hall JE. "Guyton and Hall Textbook of Medical Physiology". Elsevier. (2016). ISBN: 9781455770052
- Giangrande PLF. "Six Characters in Search of An Author: The History of the Nomenclature of Coagulation Factors". Br J Haematol. 121(5). :703-712. (2003)
- Katzung B, Trevor A. "Basic and Clinical Pharmacology". McGraw-Hill Education. (2014). ISBN: 9780071825054
- Marder VJ, Novokhatny V.. "Direct fibrinolytic agents: biochemical attributes, preclinical foundation and clinical potential.". Journal of Thrombosis and Haemostasis. (2010)
- Nazari J, Davison R, Kaplan K, Fintel D. "Adverse Reactions to Thrombolytic Agents". Med Toxicol. 2(4). :274-286. (1987)
- "Hypoplasminogenemia". https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=722. [2021-09-01]
- "RYPLAZIM® (plasminogen, human-tvmh)". https://www.fda.gov/media/149806/download
- Drebes A, de Vos M, Gill S, et al. "Prothrombin Complex Concentrates for Coagulopathy in Liver Disease: Single‐Center, Clinical Experience in 105 Patients". Hepatology Communications. (2019)
- Sorensen B, Spahn DR, Innerhofer P, Spannagl M, Rossaint R. "Clinical review: Prothrombin complex concentrates - evaluation of safety and thrombogenicity". Critical Care. 15(1). :201. (2011)
- Obaji S, Alikhan R, Rayment R, et al. "Unclassified bleeding disorders: outcome of haemostatic challenges following tranexamic acid and/or desmopressin". Haemophilia. 22(2). :285-291. (2015)
- Savage SA, Sumislawski JJ, Zarzaur BL, et al. "The new metric to define large-volume hemorrhage". Journal of Trauma and Acute Care Surgery. 78(2). :224-230. (2015)
- "The National Blood Authority’s Patient Blood Management Guideline: Module 1 – Critical Bleeding/Massive Transfusion". https://www.blood.gov.au/pbm-module-1. [2011-01-01]
- "Guidelines for Emergency Department Management of individuals with hemophilia and other bleeding disorders". https://web.archive.org/web/20250408090344/https://www.bleeding.org/healthcare-professionals/guidelines-on-care/masac-documents/masac-document-257-guidelines-for-emergency-department-management-of-individuals-with-hemophilia-and-other-bleeding-disorders. [2019-12-05]
- Müller MC, Meijers JC, Vroom MB, et al. "Utility of thromboelastography and/or thromboelastometry in adults with sepsis: a systematic review". Crit Care. 18(1). :R30. (2014)
- Müller MCA, Meijers JC, van Meenen DM, Thachil J, et al. "Thromboelastometry in critically ill patients with disseminated intravascular coagulation.". Blood Coagul Fibrinolysis. 30(5). :181-187. (2019)
- Curry NS, Davenport R, Pavord S, et al. "The use of viscoelastic haemostatic assays in the management of major bleeding". Br J Haematol. 182(6). :789-806. (2018)
- Whiting P, Al M, Westwood M, et al. "Viscoelastic point-of-care testing to assist with the diagnosis, management and monitoring of haemostasis: a systematic review and cost-effectiveness analysis". Health Technol Assess (Rockv). 19(58). :1-228. (2015)
- Wikkelsø A, Wetterslev J, Møller AM, Afshari A. "Thromboelastography (TEG) or rotational thromboelastometry (ROTEM) to monitor haemostatic treatment in bleeding patients: a systematic review with meta-analysis and trial sequential analysis". Anaesthesia. 72(4). :519-531. (2017)
- Fahrendorff M, Oliveri RS, Johansson PI. "The use of viscoelastic haemostatic assays in goal-directing treatment with allogeneic blood products – A systematic review and meta-analysis". Scandinavian Journal of Trauma, Resuscitation and Emergency Medicine. 25(1). (2017)
- Whiting D, DiNardo JA. "TEG and ROTEM: Technology and clinical applications". Am J Hematol. 89(2). :228-232. (2014)