Summary
IgA vasculitis (IgAV), previously referred to as Henoch-Schonlein purpura (HSP), is an acute immune complex-mediated small vessel vasculitis that most commonly occurs in children. Onset is often preceded by an upper respiratory tract or gastrointestinal infection; IgAV in adults may be idiopathic. Affected individuals typically develop palpable purpura, arthritis and/or arthralgia, and abdominal pain. Renal involvement (i.e., IgAV nephritis) is more common and usually more severe in adults than in children, typically manifesting with hematuria. While IgAV is a clinical diagnosis, laboratory studies are used to identify organ involvement and exclude differential diagnoses, and skin biopsy can confirm the diagnosis in patients with atypical presentations. IgAV is usually self-limited; treatment is generally supportive. Systemic glucocorticoids may be required depending on severity of manifestations (e.g., in IgAV nephritis, orchitis). IgAV has an excellent prognosis in children, usually resolving within one month when not complicated by IgAV nephritis. Adults often manifest with more severe features and have lower remission rates. All patients require follow-up assessments to rule out the development of chronic renal disease.

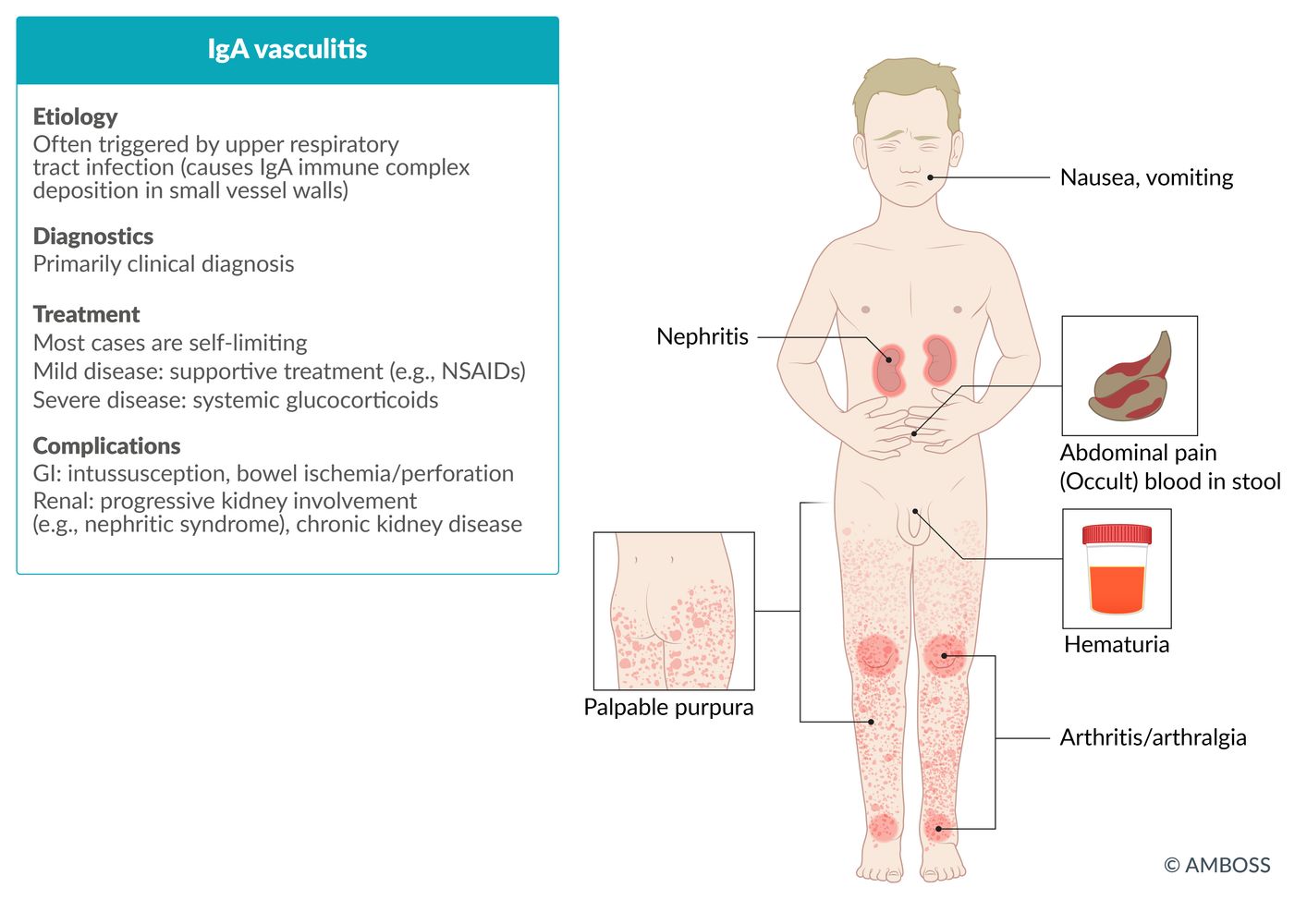
© AMBOSS
Epidemiology
- Sex: : ♂ > ♀
-
Age: more common in children
- 90% of affected individuals < 10 years [1]
- Peak incidence: 6 years [2]
Epidemiological data refers to the US, unless otherwise specified.
Etiology
The exact pathogenesis is unknown and assumed to be multifactorial. Factors that likely play a role include:
-
Preceding infection
- Up to 90% of cases are preceded by viral or bacterial infection 1–3 weeks prior. [3]
- Most commonly an upper respiratory tract infection caused by group A Streptococcus [4]
- GI infections also possible
- Many other organisms have also been associated with IgAV.
- Drugs (e.g., ACE inhibitors, clarithromycin) and vaccines
- Genetic predisposition
- Solid-organ malignancies (in adults) [5]
Pathophysiology
- Hypothesized pathophysiological mechanism: exposure to allergen/antigen (e.g., infection, drugs) → stimulation of IgA production → deposition of IgA immune complexes in vascular walls (e.g., in the skin, GI tract, joints, kidneys) → activation of complement → vascular inflammation and damage
Clinical features
An upper respiratory tract infection often precedes symptom onset by 1–3 weeks. [3]
- Constitutional symptoms: low-grade fever, malaise, fatigue
-
Skin (∼ 100% of cases) [6]
- Symmetrically distributed erythematous papules or urticarial lesions that coalesce into palpable purpura
- Bullae, pustules, and necrotic or hemorrhagic purpura (more common in adults) [5]
- Most commonly in the lower extremities, buttocks, and other areas of pressure or constraint (e.g., from clothing)
-
Joints (∼ 75% of cases) [7]
- Arthritis/arthralgia
- Most commonly in the ankles and knees
-
Gastrointestinal tract (∼ 60% of cases) [7]
- Colicky abdominal pain
- Intussusception
- Hematochezia or melena
- Nausea and/or vomiting
-
Kidneys: IgAV nephritis (20–50% of children; 50–80% of adults) ; [8]
- Nephritic syndrome
- See also “IgA nephropathy.”
-
Other organs (rare)
- Genitourinary tract (e.g., epididymo-orchitis, urethritis)
- Central and peripheral nervous system (e.g., headaches, seizures, intracerebral hemorrhage)
- Respiratory tract (e.g., mild interstitial changes, alveolar hemorrhage)
- Eyes (e.g., episcleritis, uveitis)
IgAV is characterized by PAPAH: purpura, abdominal pain, arthritis/arthralgia, and hematuria.
IgAV is an important differential diagnosis to consider in children with a limp.
IgAV is much less common but typically more severe (e.g., involving hemorrhagic or necrotic skin lesions, IgAV nephritis) in adults than in children. [8]




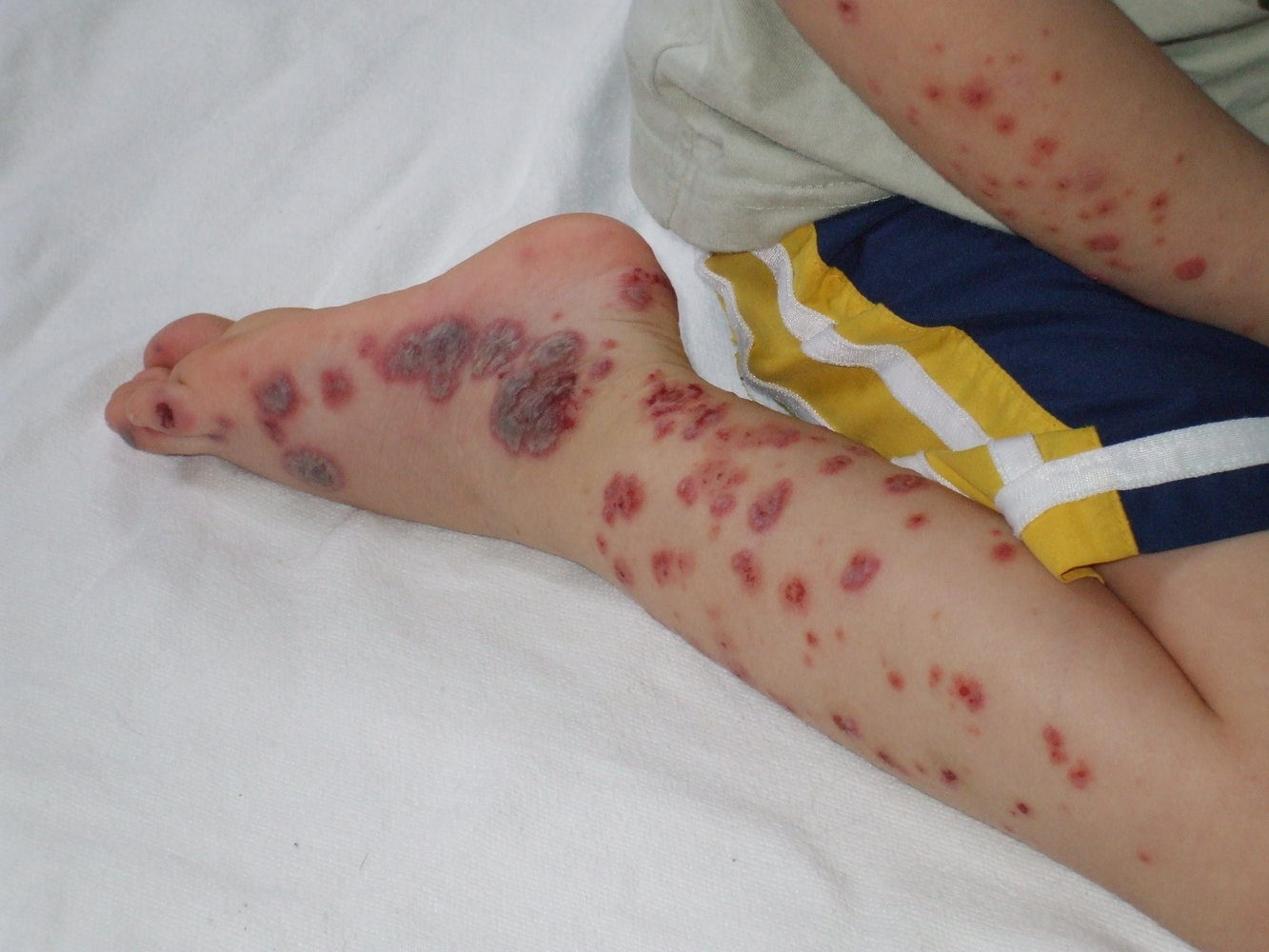
Maculopapular, reddish, non-blanching, palpable skin lesions (purpura) are seen on both the distal right arm and leg. On the lower leg, the lesions are partially confluent and present with scaling and erosion.
Source: "Purpura2", Okwikikim, Wikimedia Commons licensed under Public Domain
{kind=link}
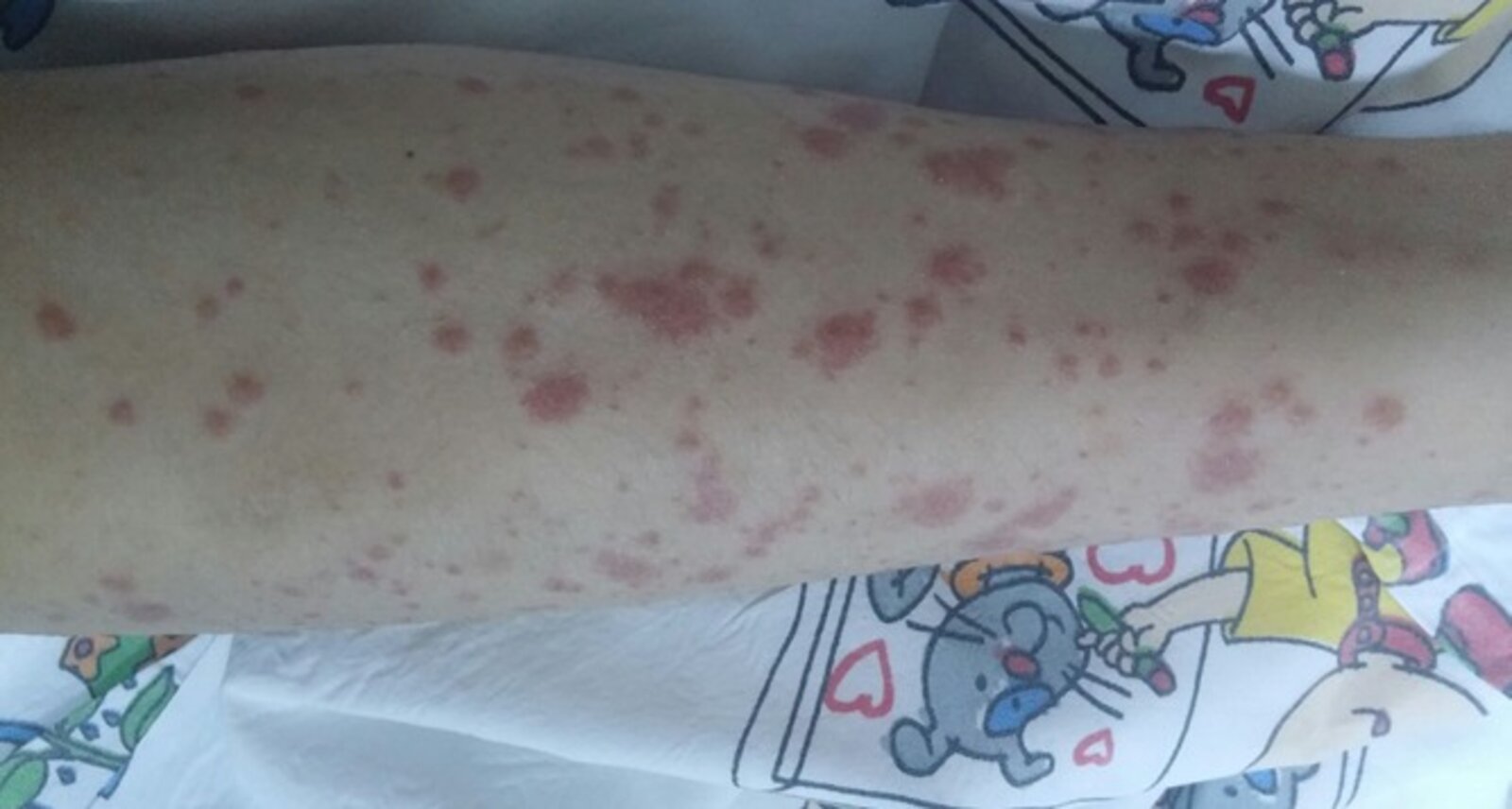
Multiple well-defined erythematous skin lesions (purpura) are distributed on the shin of this pediatric patient. Some purpura have coalesced to form ecchymoses.
Purpura may be palpable and nonblanchable on examination.
Source: “Figure 1, in: Henoch-schonlein purpura associated with primary active epstein barr virus infection: a case report” by B. Karakayali, S. Yilmaz, D. Çakir et al., The Pan African Medical Journal, licensed under CC BY 4.0.
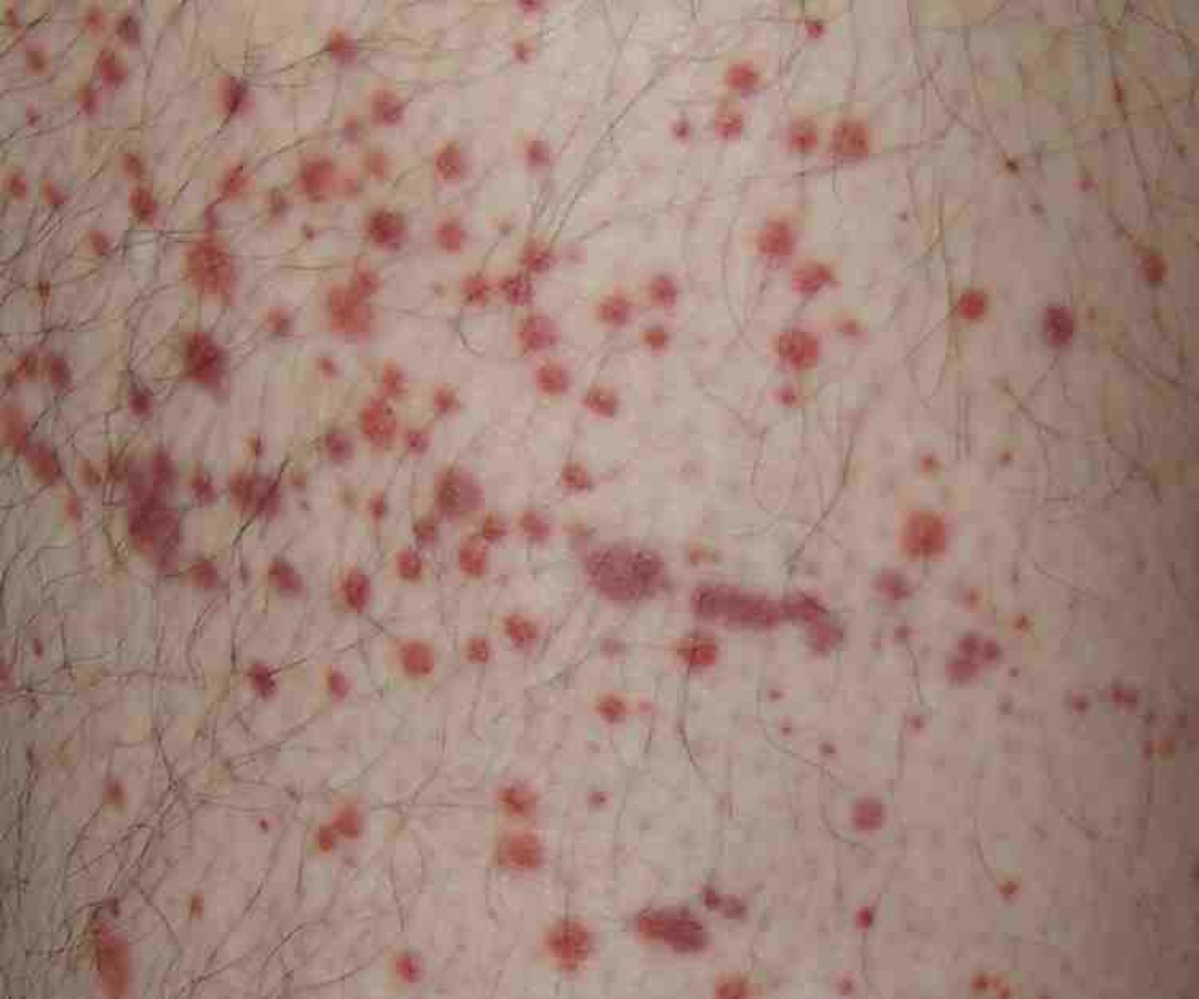
Photograph of the leg of an 18-year-old male patient
Multiple well-defined erythematous skin lesions (purpura) are visible. Some purpura have coalesced to form ecchymoses. Examination revealed the purpura to be palpable and nonblanchable.
The tetrad of purpura, arthritis, abdominal pain, and kidney impairment is typical of IgA vasculitis. Histopathology is required to confirm the diagnosis of IgA vasculitis.
Source: “Purpura” by Hektor, Wikimedia Commons, licensed under CC BY-SA 3.0.
{kind=link}
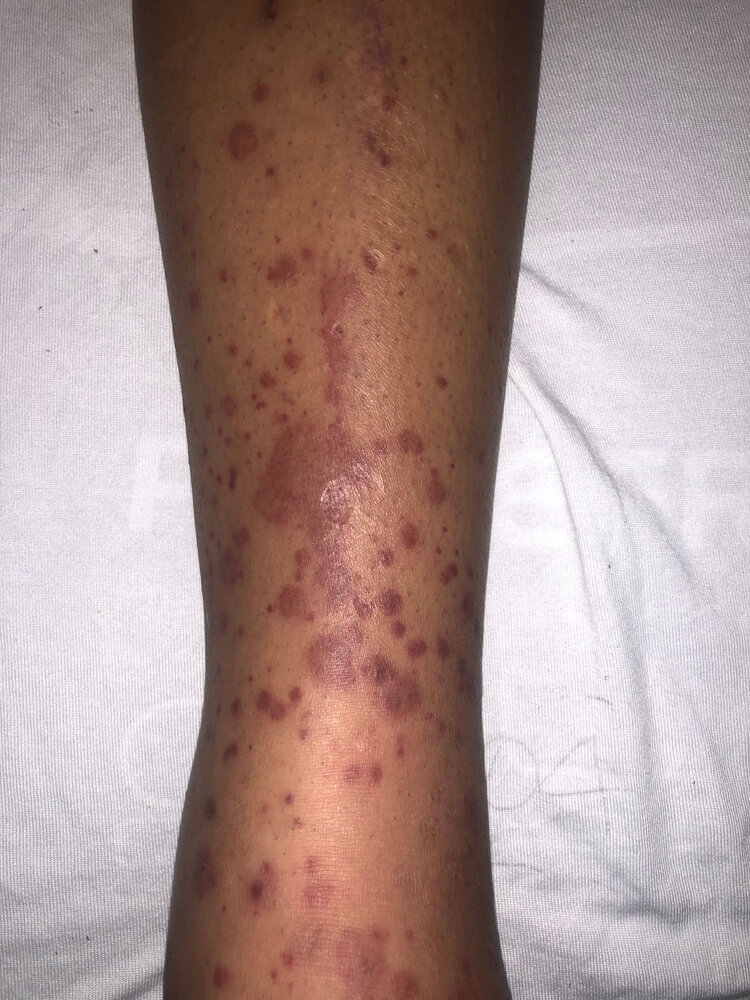
Lower extremity of a patient with painful rash
Multiple, partially confluent, erythematous skin lesions and small dark purple bumps (palpable purpura) are visible around the patient's ankle and lower leg. The rash spreads bilaterally and is accompanied by surrounding edema.
These findings are typical of IgA vasculitis (formerly Henoch-Schonlein purpura), for which the tetrad of rash, arthritis, abdominal pain, and kidney impairment is characteristic.
Source: “Figure 2, in: Henoch-Schönlein Purpura in the Adult, a Case Report” by Ivan Virovets, Danielle Biggs, jetem, licensed under CC BY 4.0.
Diagnosis
Approach [5][7][9][10]
-
Pediatric and adult patients
- Make a clinical diagnosis of IgAV based on typical clinical features.
- Check history for recent infections, medications, and vaccination as potential precipitating factors.
-
Obtain laboratory studies to identify organ involvement and exclude alternative diagnoses.
- CBC, BMP, coagulation panel
- Urinalysis and occult stool blood test
- If abdominal pain is present, obtain imaging studies to investigate for GI bleeding and intussusception.
- If the diagnosis remains unclear, refer to a specialist for confirmatory biopsy. [11]
-
Adult-specific considerations [5][10][11]
- A biopsy is typically required to confirm the diagnosis in adults.
- Consider cancer screening if the cause of IgAV is not established. [5][12]
Suspect IgA vasculitis in any patient with palpable purpura, arthralgia, and abdominal pain. [7]
Consider the differential diagnosis for palpable purpura (e.g., small-vessel vasculitides, coagulopathies).
Laboratory studies [5][7][9]
Baseline laboratory studies
Obtain the following studies in all patients to assess for renal and GI involvement:
-
CBC
- Usually normal platelet count
- ↓ Hemoglobin suggests GI bleeding.
- BMP: ↑ creatinine and/or ↑ BUN suggest IgAV nephritis
- Coagulation panel: typically normal
-
Urine: may be normal, abnormal values suggest IgAV nephritis
- Urinalysis (first sample of the day): hematuria, proteinuria
- Microscopy: nephritic sediment (e.g, RBC casts, dysmorphic RBCs)
- ↑ Urine protein-creatinine ratio
- Stool blood test: positive if there is GI bleeding
Consider an alternative diagnosis if coagulopathy and thrombocytopenia are present. [8]
Additional laboratory studies
The following studies may support the diagnosis of IgAV but are not required in the diagnostic workup.
- LFTs: ↓ albumin due to intestinal protein loss
- ↑ ESR, CRP
-
If preceding streptococcal infection:
- Hypocomplementemia [13]
- ↑ Antistreptolysin O (ASO) titers [4]
- ↑ IgA in serum: in ∼ 60% of cases; not specific for IgAV [8]
Imaging [5][9]
Imaging studies should be obtained according to the patient's symptoms and/or suspected complications.
- Abdominal ultrasound: indicated in patients with abdominal pain to evaluate for intussusception
- CT abdomen and pelvis: if ultrasound is inconclusive or if further evaluation is required
- Duplex ultrasound of the scrotum: if there is testicular pain to evaluate for orchitis or torsion
- Additional studies: based on clinical presentation (e.g., MRI brain to evaluate for cerebral vasculitis)
Intussusception is the most common complication of IgAV requiring surgery in children. [9]

Biopsy [8][9]
Histology from the skin and/or kidney confirms the diagnosis of IgAV. [8]
-
Skin
- Indications: atypical skin features
-
Findings
- Leukocytoclastic vasculitis
- IgA and C3 complex deposition (hallmark) in small vessels of the superficial dermis [3]
-
Kidney
- Indications: severe renal involvement [9][14]
-
Findings
- Mesangial IgA deposition
- C3 and fibrin
- Crescent formation in more severe cases [8]
EULAR/PRINTO/PReS classification criteria [15][16]
Classification criteria can be used to distinguish IgAV from other types of vasculitides, but should not be used as diagnostic criteria for IgAV. [9][15]
- Purpura or petechiae (predominantly in the lower limbs)
- Plus ≥ 1 of the following:
- Acute colicky abdominal pain
- Acute arthritis or arthralgias
- Proteinuria and/or hematuria
- Histopathology: cutaneous leukocytoclastic vasculitis or proliferative glomerulonephritis with predominant IgA deposits

")
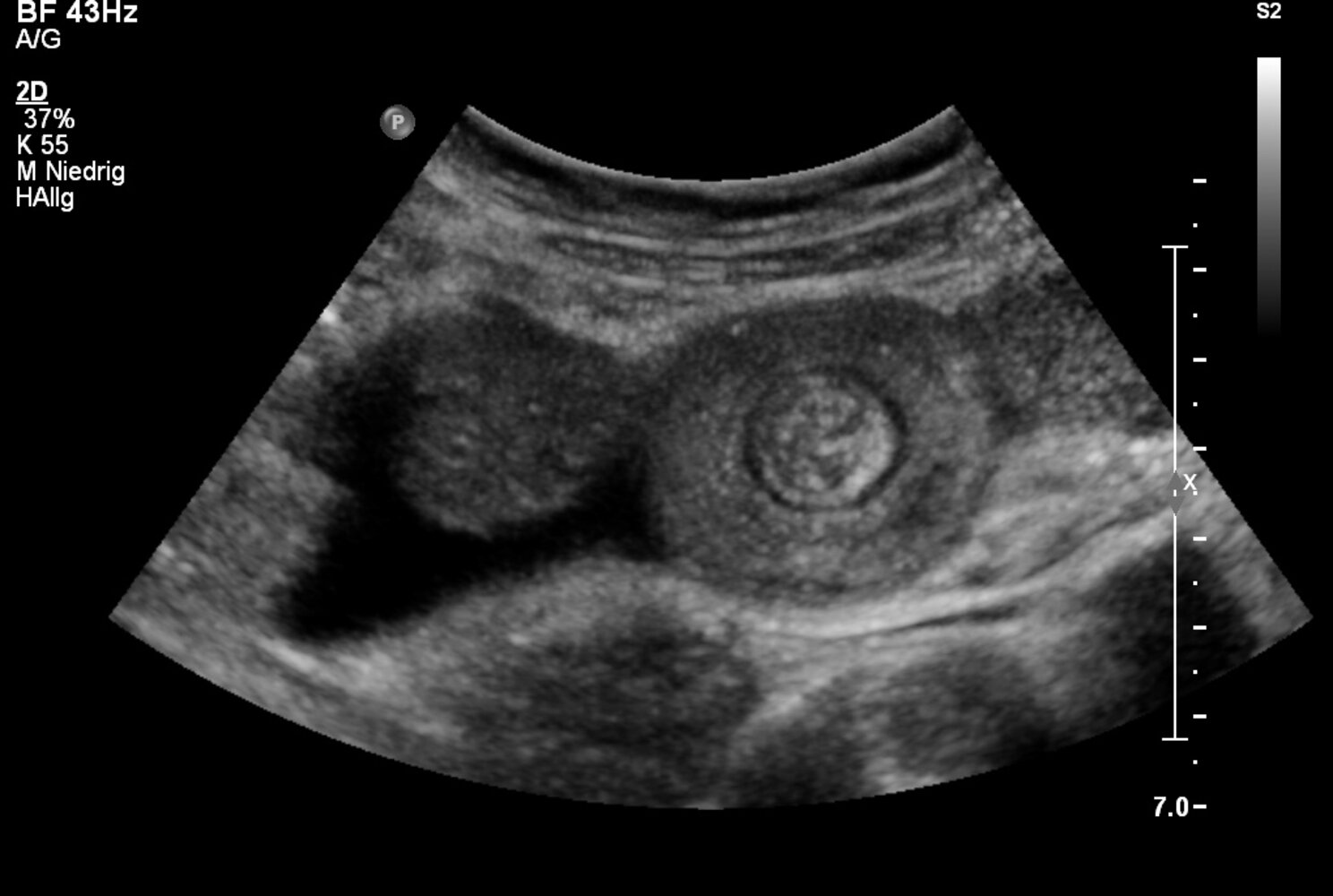
Ultrasound abdomen (bowel; transverse plane)
Concentric alternating hyperechoic and hypoechoic rings are visible. The hyperechoic rings (green overlay) are formed by mucosa and the hypoechoic rings (red overlay) by submucosa. Together the alternating layers produce a target-like appearance (target sign; bull's eye sign).
Our great thanks to Albertinen Krankenhaus, Hamburg, Germany, for kindly providing this case.
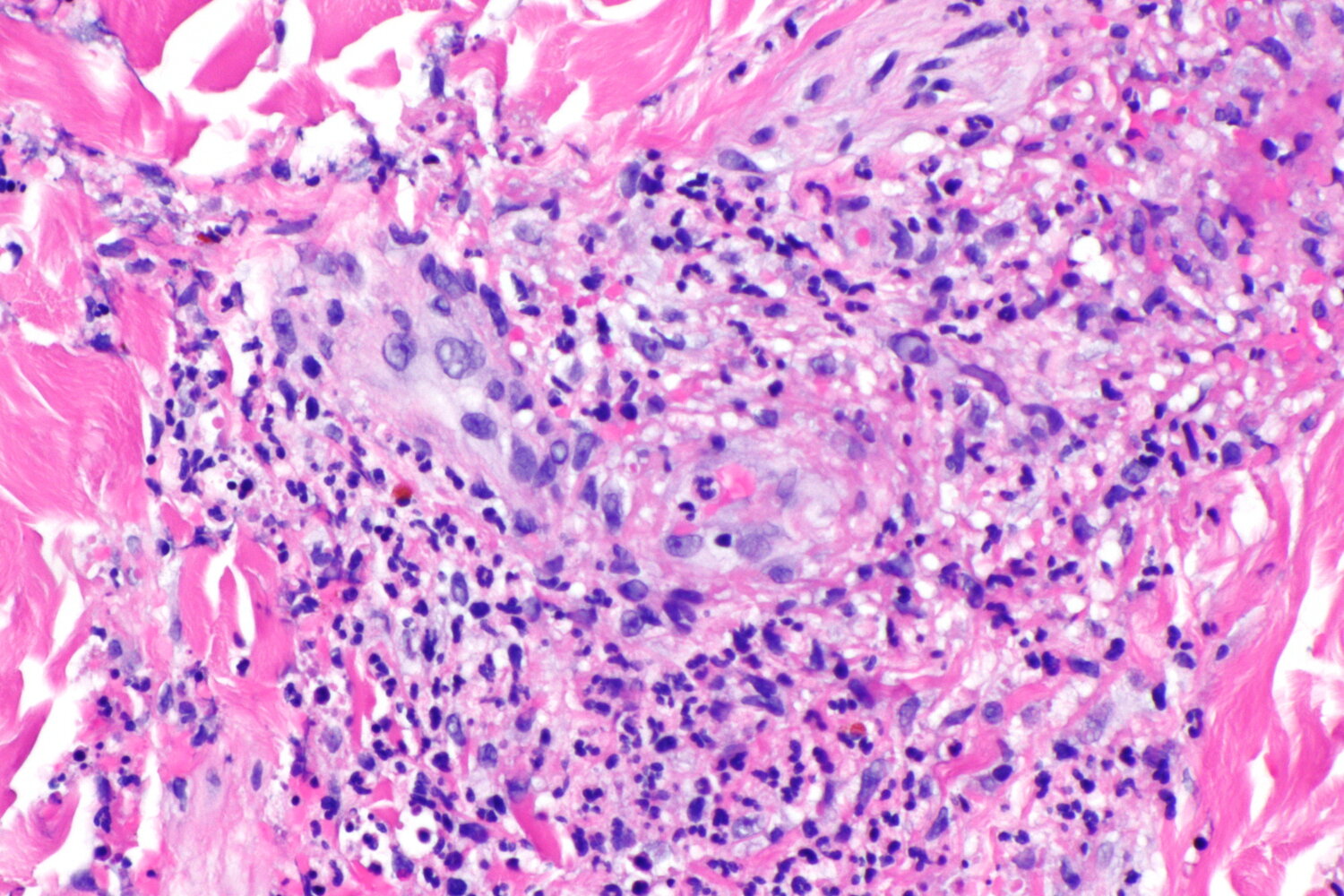
Photomicrograph of a skin tissue sample (H&E stain; high magnification)
There is focal neutrophilic inflammation (neutrophils indicated by arrowheads) neighboring a small blood vessel (marked by blue line) of the skin with the typical appearance of fragmented neutrophilic nuclei (leukocytoclasia, examples marked by arrows).
These findings are suggestive of small-vessel leukocytoclastic vasculitis (leukocytoclastic vasculitis).
Source: “Leukocytoclastic vasculitis -- high mag.jpg” by Nephron, Libre Pathology, licensed under CC BY-SA 3.0. The supplementary image with overlays of relevant areas was adapted from the image mentioned above and licensed under CC BY-SA 3.0.
{kind=link}
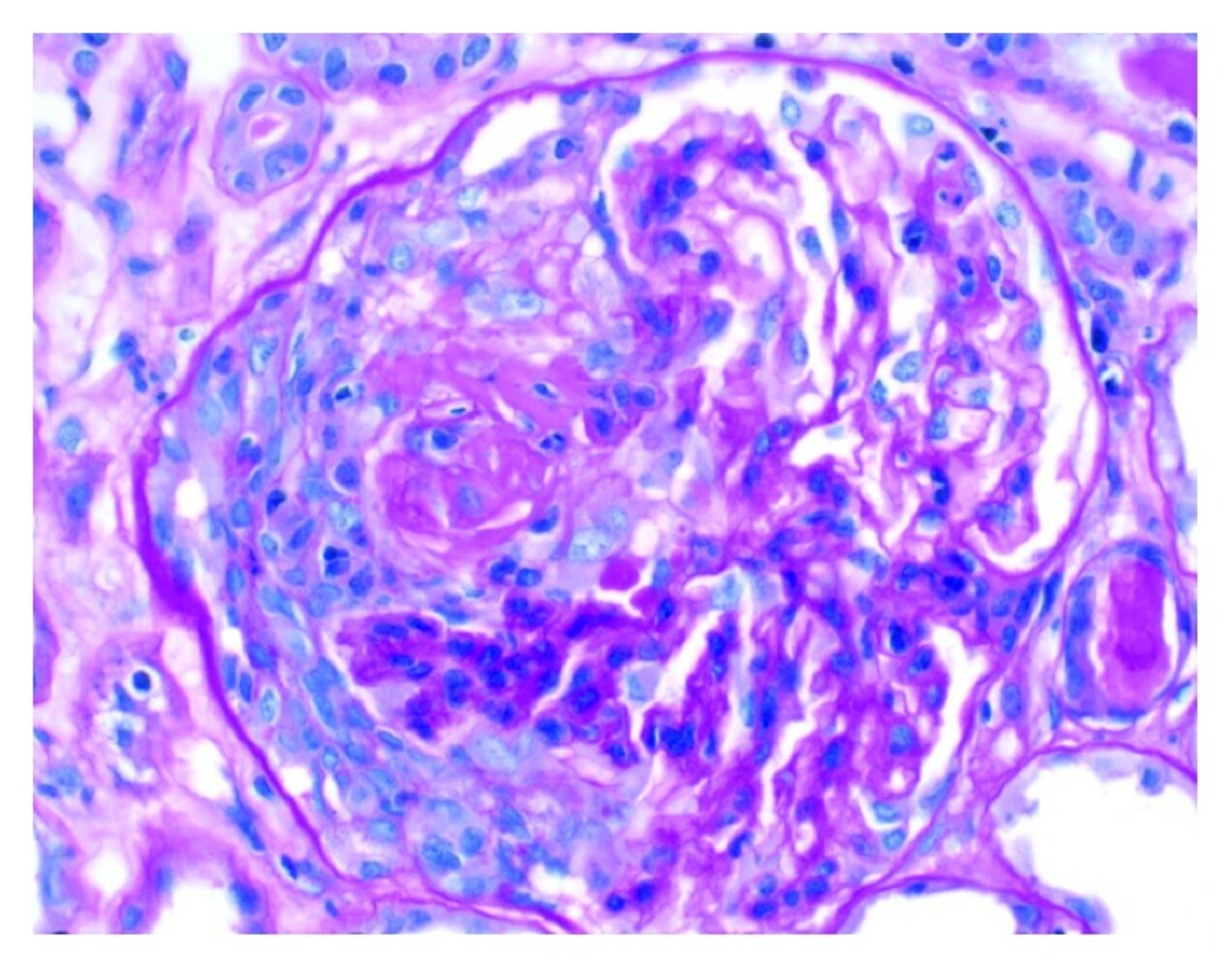
Photomicrograph of kidney biopsy tissue (H&E stain; high magnification)
A hypercellular, crescent-shaped zone with an inflammatory infiltrate (yellow overlay) is visible within the glomerulus.
Glomerular crescent formation is a characteristic histological finding in RPGN.
Source: “Figure 1, in: ANCA Vasculitis and Hemophagocytic Lymphohistiocytosis following a Fecal Microbiota Transplant” by A. Amlani, A. Bromley, A. Fifi-Mah, Hindawi - Case Reports in Rheumatology, licensed under CC BY 4.0. The supplementary image with overlays of relevant areas was adapted from the image mentioned above (© AMBOSS).
Differential diagnoses
| Differential diagnosis of IgAV based on clinical features | ||
|---|---|---|
| Clinical feature of IgAV | Differential diagnosis | Distinguishing features of the differential diagnosis |
| Purpura [17] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Arthritis/arthralgia |
|
|
|
|
|
|
|
|
| Renal disease |
|
|
IgAV is a unique cause of purpura without thrombocytopenia.
The differential diagnoses listed here are not exhaustive.
Treatment
Approach [9][10][11]
Decisions about pharmacotherapy are based on symptom severity and should be guided by a specialist.
-
All patients: Provide supportive care.
- Ensure adequate hydration: oral rehydration therapy and IV fluid therapy as needed
- Provide NSAIDs and/or acetaminophen for pain (see “Oral analgesics” for dosages).
- Treat skin lesions with leg elevation, compression stockings, and/or wound care of ulcerative lesions. [5]
- Discontinue drugs suspected of inducing IgAV. [18]
-
Patients with severe vasculitis and/or IgAV nephritis
- Initiate systemic glucocorticoid therapy.
- Consult specialists for treatment of complications, e.g., dialysis or kidney transplant for severe IgAV nephritis.
- Admit to the hospital.
Most cases of IgAV are self-limited and only require supportive care.
Avoid NSAIDs in patients with IgAV nephritis or GI bleeding. [9]
Systemic glucocorticoids [5][9]
-
Indications [9]
- IgAV nephritis
- Severe abdominal pain
- Rectal bleeding
- Orchitis
- Cerebral vasculitis
- Pulmonary hemorrhage
- Other features of severe vasculitis
-
Agents
- Mild-to-moderate disease: PO prednisone (off-label) [9][11]
- Severe disease: IV methylprednisolone (off-label) [9]
Rule out intussusception before starting systemic glucocorticoids in patients with severe abdominal pain. [5]
Specialist consultation [5][9]
Consider early consultation as follows:
- Nephrology for IgAV nephritis; treatment options include:
- Systemic glucocorticoids (first-line treatment) [9]
- Other immunosuppressive agents (second-line treatment), e.g., azathioprine, mycophenolate mofetil, IV cyclophosphamide [9]
-
RAAS inhibitors, e.g., ACEI, ARB
- Indicated in patients with persistent proteinuria (> 3 months) [9]
- See “Management of AKI" and “Management of CKD.”
- Gastroenterology: if there is blood in stool and/or severe or progressive abdominal pain
- Rheumatology: for recurrent or refractory IgAV
Disposition [19]
- All patients require scheduled outpatient follow-up to monitor for delayed renal impairment (See “Follow-up of IgAV”)
- Consider hospital admission for patients with severe IgAV if any of the following are present:
- Orchitis
- Moderate-to-severe abdominal pain
- Arthritis involving > 2 joints
- Proteinuria
- Gastrointestinal bleeding
- Inability to ambulate
Complications
-
Renal [20]
- IgAV nephritis may progress to nephrotic syndrome.
- Serious complication: rapid-progressive glomerulonephritis (RPGN) with crescent formation
- End-stage renal disease requiring dialysis or kidney transplant
-
Gastrointestinal
- Intussusception
- Small bowel infarction or perforation
We list the most important complications. The selection is not exhaustive.
Follow-up
Patients with IgAV require follow-up due to an increased risk of chronic kidney disease. [5][9][21]
-
Monitoring
- Blood pressure
- Urine studies: urinalysis, UPCR, UACR
- Serology: creatinine with eGFR
-
Frequency
- Patients with proteinuria or hypertension: every 2 weeks for 1 month, then every 1–2 months for 6–12 months, then every 3–6 months
- Patients without proteinuria or hypertension: every 2 weeks for 1 month, then every 2–3 months for 6–12 months, then annually
- Repeat if the patient develops AKI or a nonrenal flare (e.g., cutaneous lesions, GI symptoms)
-
Refer to nephrology if there is:
- Persistent hypertension
- Worsening renal function or proteinuria
- Persistent proteinuria or hematuria for > 3 months
IgAV nephritis typically develops within 6 months of IgAV symptom onset. [21]
Prognosis
- IgAV usually resolves with full recovery. However, relapse is likely in patients with previous renal involvement.
- The prognosis is worse in patients with nephrotic range proteinuria. In rare cases (∼ 1%), ESRD may occur.
External Resources
References
- Reamy BV, Williams PM, Lindsay TJ. "Henoch-Schönlein purpura". Am Fam Physician. 80(7). :697-704. (2009)
- Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR. "Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins". The Lancet. (2002)
- Lenis M. González MD Camila Krysicka Janniger MD Robert A. Schwartz MD, MPH. "Pediatric Henoch–Schönlein purpura". International Journal of Dermatology. (2009)
- Saulsbury FT. "Epidemiology of Henoch-Schönlein purpura.". Cleveland Clinic journal of medicine. 69 Suppl 2. :SII87-9. (2002)
- Kelly BG, Stratton DB, Mansour I, et al. "Navigating the initial diagnosis and management of adult IgA vasculitis: A review". JAAD Int. 8. :71-78. (2022)
- Park J, Berard RA, Grimmer J, Kirpalani A. "IgA Vasculitis: a Review and Update on the Management of Renal and Extrarenal Disease, Highlighting What’s New for Biomarkers and Treatment". Curr. Pediatr. Rep. 9(4). :118-126. (2021)
- Reamy BV, Servey JT, Williams PM. "Henoch-Schönlein Purpura (IgA Vasculitis): Rapid Evidence Review". Am Fam Physician. 102(4). :229-233. (2020)
- Pillebout E, Sunderkötter C. "IgA vasculitis". Semin Immunopathol. 43(5). :729-738. (2021)
- Ozen S, Marks SD, Brogan P, et al. "European consensus-based recommendations for diagnosis and treatment of immunoglobulin A vasculitis—the SHARE initiative". Rheumatology. 58(9). :1607-1616. (2019)
- Yaseen K, Herlitz LC, Villa-Forte A. "IgA Vasculitis in Adults: a Rare yet Challenging Disease". Curr Rheumatol Rep. 23(7). (2021)
- Walls R, Hockberger R, Gausche-Hill M, Erickson TB, Wilcox SR. "Rosen's Emergency Medicine 10th edition- Concepts and Clinical Practice E-Book". Elsevier Health Sciences. (2022). ISBN: 9780323757904
- Podjasek J, Wetter D, Pittelkow M, Wada D. "Henoch-Schönlein Purpura Associated With Solid-organ Malignancies: Three Case Reports and a Literature Review". Acta Derm Venereol. 92(4). :388-392. (2012)
- Lin Q, Min Y, Li Y, et al. "Henoch–Schönlein purpura with hypocomplementemia". Pediatr Nephrol. 27(5). :801-806. (2012)
- Rovin BH, Adler SG, Barratt J, et al. "KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases". Kidney Int. 100(4). :S1-S276. (2021)
- Ozen S, Pistorio A, Iusan SM, et al. "EULAR/PRINTO/PRES criteria for Henoch-Schonlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria". Ann Rheum Dis. 69(5). :798-806. (2010)
- Hočevar A, Rotar Z, Jurčić V, et al. "IgA vasculitis in adults: the performance of the EULAR/PRINTO/PRES classification criteria in adults". Arthritis Res Ther. 18(1). (2016)
- Dedeoglu F, Kim S. "IgA Vasculitis (Henoch-Schönlein Purpura): Clinical Manifestations and Diagnosis". UpToDate. UpToDate. https://www.uptodate.com/contents/henoch-schonlein-purpura-immunoglobulin-a-vasculitis-clinical-manifestations-and-diagnosis. [2015-12-15]
- Rasmussen C, Tisseyre M, Garon-Czmil J, et al. "Drug-induced IgA vasculitis in children and adults: Revisiting drug causality using a dual pharmacovigilance-based approach". Autoimmun Rev. 20(1). :102707. (2021)
- Masarweh K, Horovitz Y, Avital A, Spiegel R. "Establishing hospital admission criteria of pediatric Henoch–Schonlein purpura". Rheumatol Int. 34(11). :1497-1503. (2014)
- Watson L, Richardson ARW, Holt RCL, Jones CA, Beresford MW. "Henoch Schonlein Purpura – A 5-Year Review and Proposed Pathway". PLoS ONE. 7(1). :e29512. (2012)
- Sharma A. "Textbook of Systemic Vasculitis". Jaypee Brothers Medical Publishers. (2015). ISBN: 9789351526520