CME information and disclosures
To see contributor disclosures related to this article, hover over this reference: [1]
Physicians may earn CME/MOC credit by searching for an answer to a clinical question on our platform, reading content in this article that addresses that question, and completing an evaluation in which they report the question and the impact of what has been learned on clinical practice.
AMBOSS designates this Internet point-of-care activity for a maximum of 0.5 AMA PRA Category 1 Credit(s)™. Physicians should claim only credit commensurate with the extent of their participation in the activity.
For answers to questions about AMBOSS CME, including how to redeem CME/MOC credit, see "Tips and Links" at the bottom of this article.
Summary
Immune thrombocytopenia (ITP) is a type of thrombocytopenia involving the formation of autoantibodies against platelets. ITP may be a primary disease or occur secondary to a known trigger (e.g., SLE, HIV, hepatitis C, medications). It is commonly seen in children as a self-limiting illness following a viral infection, and in adults as a chronic illness. Most patients are asymptomatic, however, patients may occasionally present with minor mucocutaneous bleeding (e.g., petechiae, purpura, epistaxis) or, rarely, with severe bleeding (e.g., gastrointestinal or intracranial hemorrhage). Treatment recommendations vary depending on the presence and severity of symptoms. First-line medical therapy consists of corticosteroids, IVIG, or anti-D immunoglobulin, and is indicated for patients with non-life-threatening symptoms affecting their quality-of-life, and for adults with little-to-no symptoms and platelet counts below 30,000/mm3. Children with little-to-no symptoms can typically be managed with observation alone regardless of platelet count. Second-line treatments (i.e., thrombopoietin receptor agonists, rituximab, splenectomy) may be required for refractory, persistent or chronic cases. Patients with life-threatening hemorrhage, neurological symptoms, or those requiring urgent surgical interventions should receive immediate combination medical therapy (i.e., corticosteroids plus IVIG) along with platelet transfusions and hemostatic control interventions when necessary.
See also “Thrombocytopenia.”
Definitions
Immune thrombocytopenia has previously been referred to as idiopathic thrombocytopenic purpura, however, this term is outdated.
- Primary immune thrombocytopenia: : an autoimmune disorder characterized by isolated thrombocytopenia (< 100,000/mm3) with no known precipitating cause [2]
- Secondary immune thrombocytopenia: an autoimmune hematologic disorder causing isolated thrombocytopenia that is secondary to an identifiable trigger (see “Etiology”).
- Newly diagnosed ITP: all cases within the first 3 months of diagnosis [2]
- Persistent ITP: ITP lasting 3–12 months
- Chronic ITP: ITP lasting > 12 months
Epidemiology
- Prevalence: up to ∼ 10 per 100,000 [3][4]
- ♀ > ♂ [3]
-
Children
- Highest prevalence in children < 5 years of age [3]
- Typically self-limiting after a viral infection; 80% of cases resolve within 12 months [5]
- Adults
- Highest prevalence in individuals > 55 years of age [3]
- 80% of patients develop chronic ITP. [5]
- An incidental finding on a routine CBC in 25% of cases [6]
Epidemiological data refers to the US, unless otherwise specified.
Etiology
- Primary ITP: idiopathic (most common) [7]
-
Secondary ITP associated with:
- Autoimmune disorders: SLE, antiphospholipid syndrome
- Malignancy: lymphoma, leukemia (particularly CLL)
- Infection: HIV, HCV
- Drugs: e.g., quinine,; beta-lactam antibiotics, carbamazepine, heparin, vaccines, linezolid, sulfonamides, vancomycin, TMP-SMX, antiepileptics [8]
Pathophysiology
Antiplatelet antibodies (mostly IgG directed against, e.g., GpIIb/IIIa, GpIb/IX) bind to surface proteins on platelets → sequestration by spleen and liver → ↓ platelet count → bone marrow megakaryocytes and platelet production increase in response (in most cases) [9]
Clinical features
Clinical features can correlate with platelet count (see also “Clinical features of thrombocytopenia” and “Clinical features of bleeding disorders”).
-
Most commonly
- Asymptomatic
- Splenomegaly is typically absent. [10]
-
Minor mucocutaneous bleeding (less common)
- Subcutaneous: e.g., bruising, petechiae, purpura
- Mucosal: e.g., mild epistaxis, gingival bleeding
-
Other types of bleeding (rare)
- Gastrointestinal: e.g, melena
- Genitourinary: e.g., hematuria, menorrhagia
- CNS: e.g., features of intracranial hemorrhage
- Prolonged or excessive traumatic or surgical bleeding
Splenomegaly is very unusual in ITP and makes other diagnoses more likely!
There should be suspicion for ITP in a child with thrombocytopenia and petechiae following a viral illness!




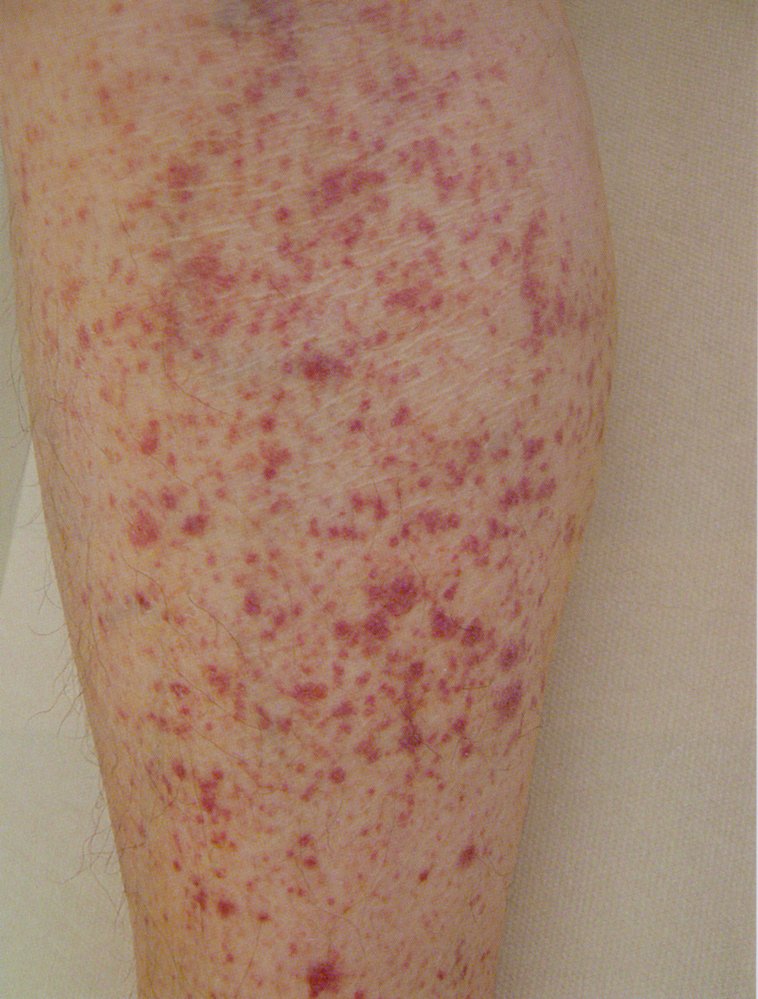
The lower leg displays numerous red spots, each about the size of a pinhead, that do not blanch or disappear when pressed. These are indicative of petechial hemorrhages related to thrombocytopenia.
Source: © IMPP
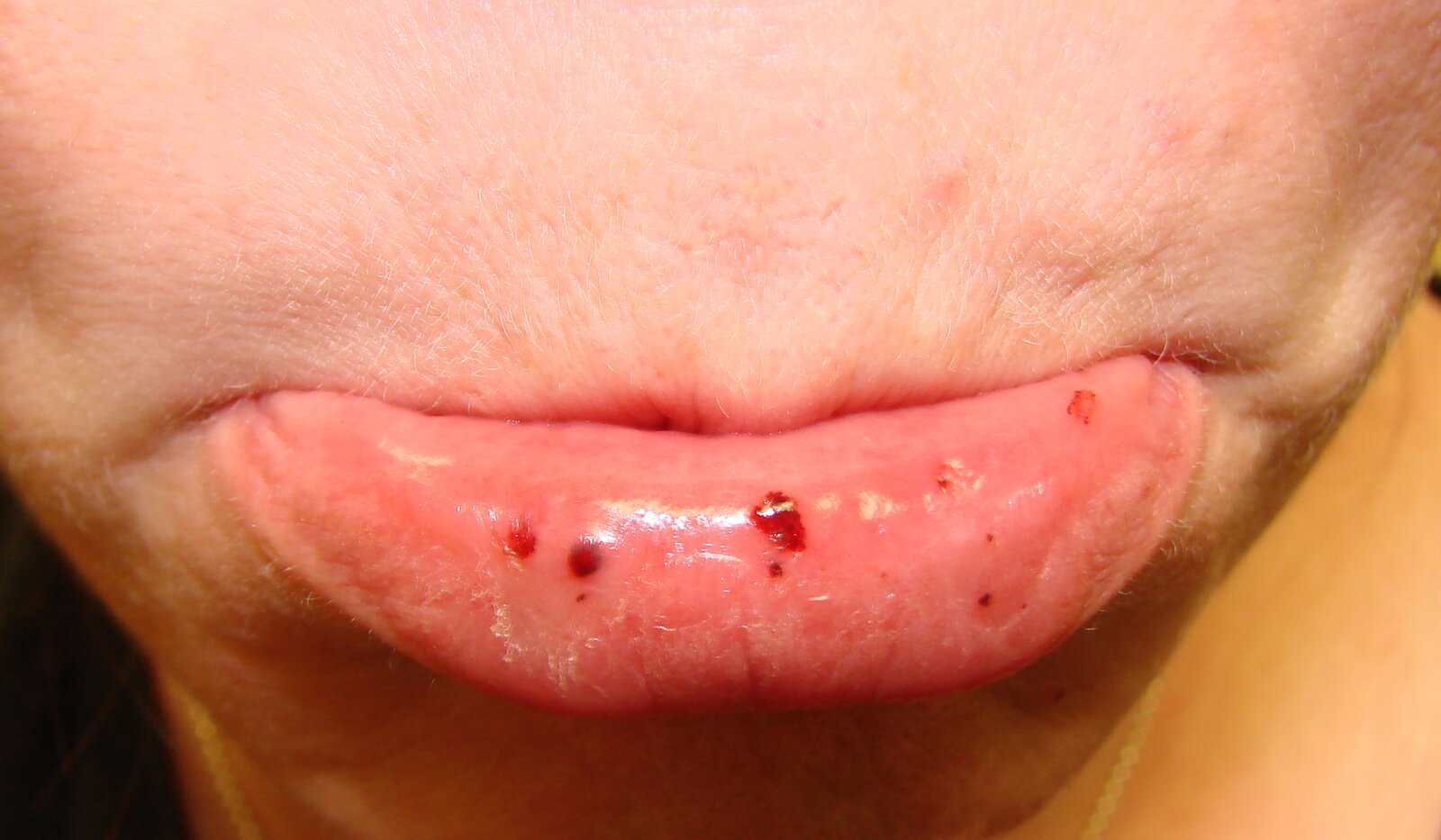
Lower lip of a person with immune thrombocytopenia:
Several small, punctiform hemorrhages (petechiae) can be seen on the oral mucosa of the lower lip. This finding is consistent with the diagnosis of immune thrombocytopenia (ITP).
Source: “Oral petechiae.JPG” by Mdscottis, Wikimedia Commons, licensed under CC BY-SA 3.0.
{kind=link}
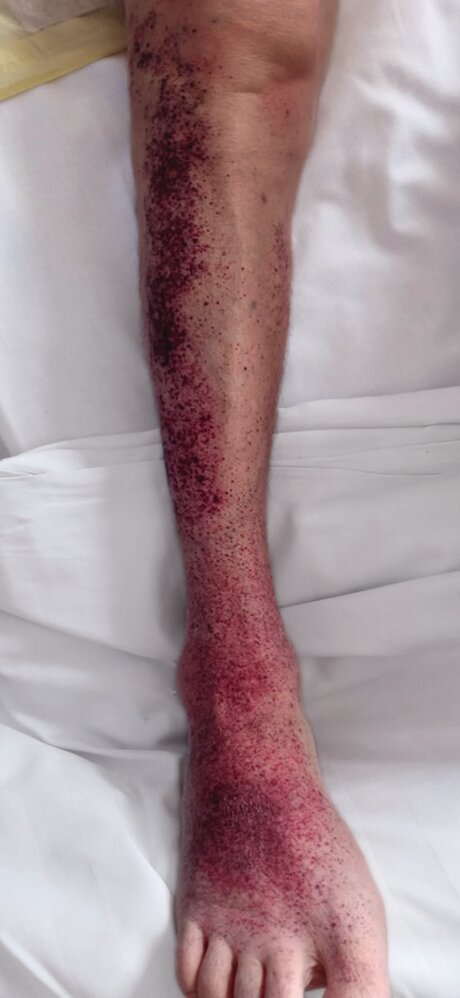
Left lower leg examination of a 54-year-old female patient with immune thrombocytopenia:
The dorsum of the foot and the outer aspect of the lower leg show multiple punctate, non-blanching hemorrhages. These lesions range in color from erythematous to livid and may occasionally merge to form larger areas of hemorrhagic skin, known as purpura. This finding is characteristic of petechial skin hemorrhages associated with thrombocytopenia.
Source: © IMPP
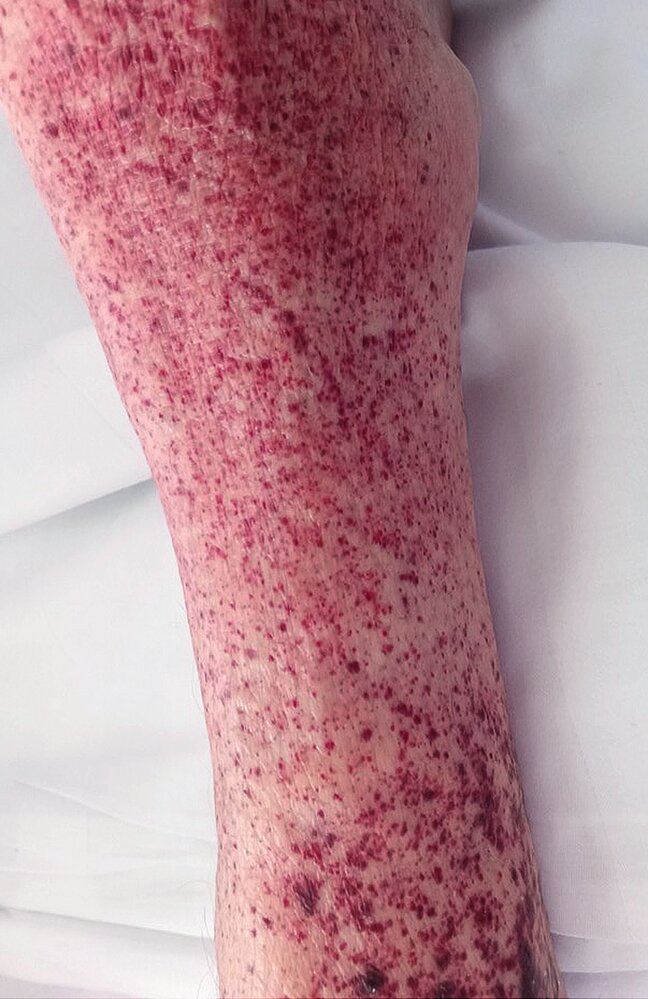
Lower leg examination of a 54-year-old female patient with immune thrombocytopenia:
The lower leg exhibits multiple pinhead-sized, dark red to purple hemorrhages on the skin. These changes, which cannot be blanched or pushed away, are distributed throughout the entire lower leg, a condition known as purpura. This finding is characteristic of petechial skin hemorrhages, commonly associated with thrombocytopenia.
Source: © IMPP
Diagnosis
ITP is a diagnosis of exclusion; patients typically have a low platelet count with no other abnormalities. [5]
Laboratory studies [10][11][12]
Routine
- CBC: ↓ platelet count (< 100,000/mm3)
- Coagulation panel: usually normal
- Bleeding time: may be prolonged
- Peripheral blood smear: normal to large platelets [13]
Additional investigations
- All adults: HIV and HCV screening
-
Bone marrow biopsy
- Consider in atypical cases or if there is diagnostic uncertainty
- Findings: normal or ↑ megakaryocytes [5]
- Additional testing as required: consider in suspected secondary ITP, e.g., antinuclear antibodies in SLE or H. pylori testing if the patient has GI symptoms or is from a high prevalence area
Differential diagnoses
See “Differential diagnosis of platelet disorders.”
The differential diagnoses listed here are not exhaustive.
Treatment
Approach
These recommendations are consistent with the 2019 American Society of Hematology ITP guidelines and the 2019 International Consensus Report on primary ITP investigation and management. [14][15]
| Management approach for ITP [14][15] | ||
|---|---|---|
| Management | Newly diagnosed ITP | Persistent or chronic ITP |
| All patients |
|
|
Patients requiring emergency treatment:
|
|
|
Patients with:
|
|
|
| Patients with: Significant non-life-threatening mucosal bleeding Symptoms impacting quality-of-life |
|
|
| Disposition |
|
|
Patients that can be managed as outpatients should receive expedited hematology follow-up within 24–72 hours. [14]
Conservative management [14][15]
-
Indications for observation
- Children: no symptoms or only mild mucocutaneous bleeding with any platelet count
- Adults: no symptoms or minor mucocutaneous bleeding with a platelet count of ≥ 30,000/mm3
- All patients: Refer to hematology for regular monitoring and counseling on bleeding risks.
First-line medical therapy [14][15]
-
Preferred: Corticosteroids [17]
- Dexamethasone [14]
- OR prednisone [14][15]
-
Alternatives: if contraindication, non-response, or intolerance to corticosteroids
- Intravenous immunoglobulin (IVIG)
- OR anti-Rho(D) immunoglobulin
- FDA black box warning: risk of intravascular hemolysis
- In-hospital monitoring: recommended for at least 8 hours after administration
- Urinalysis for hematuria and hemoglobinuria prior to therapy, and at 2 hours and 4 hours after administration.
- Order hemolysis workup in patients with characteristic clinical features (e.g., back pain, chills, urine discoloration)
- Side effects [17]
- IVIG: e.g., thrombosis, aseptic meningitis, infusion reactions
- Anti-Rho(D) immunoglobulin: e.g., intravascular hemolysis that can cause anemia, multiorgan dysfunction, AKI, renal failure, DIC, or death
Anti-Rho(D) immunoglobulin can cause potentially fatal intravascular hemolysis in patients with ITP. Close monitoring is recommended.
Subsequent therapeutic options [15]
The following options should be considered in consultation with a specialist for patients with newly-diagnosed ITP refractory to first-line medical therapy, or persistent/chronic ITP.
-
Thrombopoietin receptor agonists (TPO-RAs)
- Romiplostim
- Eltrombopag
- Avatrombopag
- Rituximab [2]
-
Splenectomy
-
Indications
- Treatment-resistant thrombocytopenia lasting > 12 months
- Severe bleeding that does not respond to any other treatment
- Procedure: Minimally invasive laparoscopic surgery is preferred.
- Complications: See “Asplenic sepsis.”
- Prevention of complications: See “Management of asplenic patients.”
-
Indications
External Resources
References
- "Contributor Disclosures - Immune thrombocytopenia. All of the relevant financial relationships listed for the following individuals have been mitigated: Jan Schlebes (medical editor, is a shareholder in Fresenius SE & Co KGaA). None of the other individuals in control of the content for this article reported relevant financial relationships with ineligible companies. For details, please review our full conflict of interest (COI) policy:"
- Kistangari G, McCrae KR. "Immune thrombocytopenia.". Hematol Oncol Clin North Am. 27(3). :495-520. (2013)
- Neunert C, Lim W, Crowther M, et al. "The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia". 117. 16(4190-4207). (2011)
- Jameson JL, Fauci AS, Kasper DL, et al. "Harrison's Principles of Internal Medicine, Twentieth Edition (Vol.1 & Vol.2)". McGraw-Hill Education / Medical. (2018). ISBN: 9781259644030
- Silberstein LE, Anastasi J. "Hematology: Basic Principles and Practice E-Book". Elsevier Health Sciences. (2017). ISBN: 9780323509718
- D'Andrea G, Chetta M, Margaglione M. "Inherited platelet disorders: thrombocytopenias and thrombocytopathies.". Blood Transfus. 7(4). :278-92. (2009)
- Neunert C, et al. "American Society of Hematology 2019 guidelines for immune thrombocytopenia". Blood Adv. 3(23). :3829-3866. (2019)
- Provan D, Arnold DM, Bussel JB, et al. "Updated international consensus report on the investigation and management of primary immune thrombocytopenia". Blood Adv. 3(22). :3780-3817. (2019)
- Izak M, Bussel JB. "Management of thrombocytopenia". F1000Prime Rep. 6. (2014)
- Estcourt LJ, Birchall J, Allard S, et al. "Guidelines for the use of platelet transfusions". Br J Haematol. 176(3). :365-394. (2016)
- Rodeghiero F, Stasi R, Gernsheimer T, et al. "Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group". Blood. 113(11). :2386-2393. (2009)
- Cines DB, Bussel JB, Liebman HA, Luning Prak ET. "The ITP syndrome: pathogenic and clinical diversity". Blood. 113(26). :6511-6521. (2009)
- Mitta A, Curtis BR, Reese JA, George JN. "Drug‐Induced Thrombocytopenia: 2019 Update of Clinical and Laboratory Data". Am J Hematol. 94(3). :E76-E78. (2018)
- SEGAL JB, POWE NR. "Prevalence of immune thrombocytopenia: analyses of administrative data". J Thromb Haemost. 4(11). :2377-2383. (2006)
- Terrell DR, Beebe LA, Vesely SK, et al. "The incidence of immune thrombocytopenic purpura in children and adults: A critical review of published reports.". Am J Hematol. 85(3). :174-80. (2010)
- Caligiuri M, Levi MM, Kaushansky K, et al. "Williams Hematology, 9E". McGraw-Hill Education / Medical. (2015). ISBN: 9780071833004
- Cines DB, Blanchette VS. "Immune Thrombocytopenic Purpura". N Engl J Med. 346(13). :995-1008. (2002)