Summary
Interstitial lung diseases (ILDs) are a diverse group of rare, highly morbid pulmonary disorders characterized by inflammation and progressive scarring (fibrosis) of the lungs. The most common types of ILD are idiopathic pulmonary fibrosis (IPF), connective tissue disease-associated ILD (CTD-ILD), chronic hypersensitivity pneumonitis, and smoking-related ILD. Cough and progressive exertional dyspnea are the most common symptoms. Bibasilar inspiratory crackles or rales are typically heard on auscultation. While most forms of ILD have similar clinical features, different types of ILD have unique epidemiological and radiographic features. A multidisciplinary approach is ideal for distinguishing IPF from other types of ILD, based on a thorough medical history and HRCT findings. Bronchoscopy or surgical lung biopsy is sometimes necessary to confirm the diagnosis. Smoking cessation, removal of exposures, symptom management, and advance care planning are central to the management of all forms of ILD. Long-term treatment of IPF may include antifibrotic agents. Corticosteroids should only be used to treat select cases of acute exacerbations of IPF. For other causes of ILD, treatment may involve systemic immunomodulators targeting the underlying cause (e.g., rheumatoid arthritis). Advanced stages of ILD can result in pulmonary hypertension and right heart failure. Lung transplantation is the only curative treatment.
Definitions
-
Interstitial lung diseases (ILDs)
- A term encompassing a heterogeneous group of disorders characterized by inflammation and progressive fibrosis of the lungs
- Cough and progressive exertional dyspnea are the most common symptoms [1]
- Idiopathic interstitial pneumonias (IIPs): a group of interstitial lung diseases of unknown cause, also described as diffuse parenchymal lung diseases (DPLDs), characterized by inflammation and fibrosis of the pulmonary interstitium but frequently also affect the airspaces, peripheral airways, and vessels
- Pneumoconioses: a group of restrictive interstitial lung diseases caused by the inhalation of certain substances (mainly dusts) that often affect miners and agricultural workers
Etiology
Idiopathic interstitial pneumonias (IIPs)
- Idiopathic pulmonary fibrosis (IPF)
-
Other IIPs
- Acute interstitial pneumonia (AIP)
- Cryptogenic organizing pneumonia (COP)
- Respiratory bronchiolitis-associated interstitial lung disease (RB-ILD)
- Desquamative interstitial pneumonia (DIP)
- Nonspecific interstitial pneumonia (NSIP)
- Lymphoid interstitial pneumonia (LIP)
- Idiopathic pleuroparenchymal fibroelastosis
Secondary to a known cause
Exposure-related (environmental and occupational)
-
Hypersensitivity pneumonitis (extrinsic allergic alveolitis)
- Farmer's lung
- Pigeon breeder's lung
- Chemical worker's lung
-
Pneumoconioses
- Coal worker's lung
- Asbestosis
- Silicosis
- Berylliosis
- Radiation pneumonitis
-
Substance-induced ILD
-
Chemotherapeutic agents
- Bleomycin
- Busulfan
- Methotrexate
- Other medications
- Amiodarone
- Nitrofurantoin (see “Nitrofurantoin-induced lung disease”)
- Phenytoin
- Penicillamine
- Other substances
- Cocaine
- Heroin
-
Chemotherapeutic agents
Secondary to underlying disease
-
Connective tissue diseases
- Systemic sclerosis
- Polymyositis-dermatomyositis
- Rheumatoid arthritis
- Systemic lupus erythematosus
- Mixed connective tissue disease
- Granulomatous diseases: sarcoidosis
-
Vasculitis
- Granulomatosis with polyangiitis
- Eosinophilic granulomatosis with polyangiitis
- Hypersensitivity reactions: eosinophilic pneumonia
-
Alveolar filling diseases
- Anti-glomerular basement membrane antibody (Goodpasture syndrome)
- Idiopathic pulmonary hemosiderosis
- Pulmonary alveolar proteinosis
-
Infectious diseases
- Tuberculosis
- Legionellosis
-
Miscellaneous
- Pulmonary Langerhans cell histiocytosis
- Amyloidosis
- Lymphangioleiomyomatosis (LAM)
Classification of idiopathic interstitial pneumonias
Idiopathic pulmonary fibrosis (IPF)
- Definition: most common type of ILD, characterized by irreversible pulmonary fibrosis and impaired pulmonary function
-
Epidemiology
- Incidence: 10:100,000 cases per year [2]
- Affects mostly men 50–70 years of age
-
Diagnosis [3]
- Requires the absence of other known causes of interstitial lung disease (e.g., medication, environmental exposures, CTD-ILD)
-
Presence of usual interstitial pneumonia (UIP) pattern on HRCT or histopathologic studies
- Honeycomb appearance with or without traction bronchiectasis
- Ground-glass opacification with superimposed reticular abnormalities
- Bibasal subpleural distribution
- Prognosis: Respiratory failure usually occurs within 3–7 years.
Acute interstitial pneumonia (AIP)
- Definition: a severe, acute ILD that can rapidly progress to respiratory failure
- Epidemiology: most commonly affects individuals without preexisting lung conditions
- Diagnostics: histologically characterized by diffuse alveolar damage
Cryptogenic organizing pneumonia (COP)
- Definition: a rare, type of ILD characterized by inflammation of the bronchioles, alveolar ducts, and alveolar walls
-
Epidemiology
- Incidence: 1–3 per 100,000 hospital admissions [4]
- Affects mostly individuals 40–50 years of age
- Diagnostics: histologically characterized by the presence of Masson bodies (granulation tissue buds made of foamy macrophages, mononuclear cells, and fibrous tissue) and chronic patchy interstitial inflammation without fibrosis
Nonspecific interstitial pneumonia (NSIP)
- Definition: a type of ILD characterized by a mild to moderate chronic interstitial inflammation, without specific histopathologic findings that characterize UIP
- Epidemiology: affects nonsmoker women 50–60 years of age
- Etiology: associated with connective tissue diseases (e.g., systemic sclerosis), HIV infection, and hypersensitivity pneumonitis
-
Diagnostics
- Immediate subpleural sparing on imaging studies is considered specific for NSIP.
- Histological findings include interstitial thickening due to fibrosis and/or inflammatory cells
")
Desquamative interstitial pneumonia (DIP)
- Definition: a rare type of ILD characterized by lung inflammation due to intraalveolar mononuclear infiltration
- Epidemiology: affects men 40–50 years of age with a history of smoking
-
Diagnostics
- Imaging studies show ground-glass opacities in the lower pulmonary lobes, usually without peripheral reticular opacities
- Histologically characterized by intraalveolar accumulation of macrophages and thickening of alveolar septa
Respiratory bronchiolitis-interstitial lung disease (RB-ILD)
- Definition: a rare, mild ILD characterized by bronchiolar inflammation
- Epidemiology: affects individuals 30–50 years of age with a history of smoking
- Diagnostics: histologically characterized by accumulation of brown pigmented macrophages and bronchiolar submucosal inflammation
Lymphocytic interstitial pneumonia (LIP)
- Definition: a rare ILD characterized by lymphocytic infiltration of the alveolar and alveolar septa
- Epidemiology: affects adults (especially women) of all ages
- Etiology: Associated with autoimmune (e.g., Sjögren disease, SLE) disorders, lymphoproliferative disorders, and HIV infection
- Diagnostics: histologically characterized by diffuse alveolar and interstitial infiltration with plasma cells and polyclonal lymphocytes
Idiopathic pleuroparenchymal fibroelastosis (IPPFE)
- Definition: a rare ILD characterized by pleural and subpleural fibrosis
- Epidemiology: affects nonsmoker individuals between 50 and 60 years of age
-
Diagnostics
- Imaging studies show pleural and subpleural thickening of the upper pulmonary lobes on imaging studies
- Histological findings include intraalveolar fibrosis and elastosis of the alveolar walls
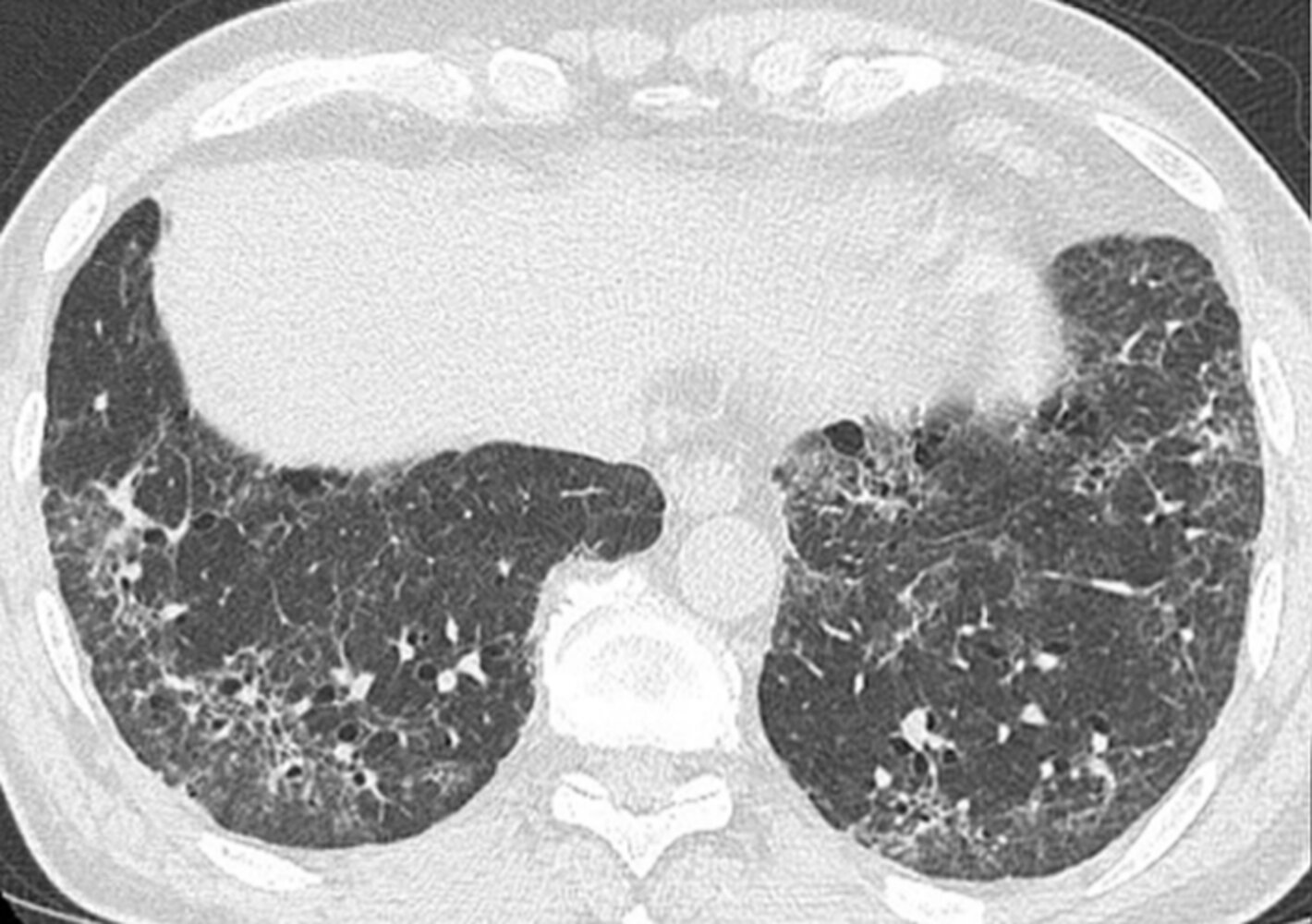
CT chest (axial plane; lung window)
A fine reticular pattern (example indicated by red outline) is accompanied by traction bronchiectasis (examples indicated by arrowheads) and mild ground-glass opacification (example indicated by green outline). There is some sparing of the immediate subpleural regions and no significant honeycombing. The appearance is consistent with the diagnosis of NSIP.
NSIP classically presents as a lower lung predominant relatively symmetric bilateral interstitial lung disease with little to no honeycombing. Although sometimes idiopathic in origin, NSIP is often associated with connective tissue disease.
Imaging features of the cellular and fibrotic subtypes of NSIP can overlap; however, the dominant feature of the cellular subtype is ground-glass opacification, whereas a common feature of the fibrotic subtype is traction bronchiectasis. A reticular pattern is seen in many fibrotic diseases, among them usual interstitial pneumonia (UIP), sarcoidosis, and hypersensitivity pneumonitis.
Source: “Fig 1E, In: Clinical features of acute fibrinous and organizing pneumonia: An early histologic pattern of various acute inflammatory lung diseases” by Onishi Y, Kawamura T, Higashino T, Mimura R, Tsukamoto H, Sasaki S, PLOS ONE, licensed under CC BY 4.0. Modifications: image cropped and removal of the letter E. The supplementary image with overlays of relevant areas was adapted from the image mentioned above (© AMBOSS).
Overview of pneumoconioses
| Overview of pneumoconioses [5][6] | ||||
|---|---|---|---|---|
| Type | Etiology | Population at risk | Clinical features | Chest x-ray |
| Asbestosis [7][8] |
|
|
|
|
| Silicosis |
|
|
|
|
| Aluminosis [9][10] |
|
|
|
|
| Anthracosis [11][12] |
|
|
|
|
| Coal workers' pneumoconiosis [11][12] |
|
|
|
|
| Berylliosis |
|
|
|
|
| Pulmonary siderosis [15][16] |
|
|
|
|
| Organic dust toxic syndrome |
|
|
|
|
| Byssinosis [17] |
|
|
|
|
Although coal is mined from under the earth, the upper lobes of the lungs are primarily affected.




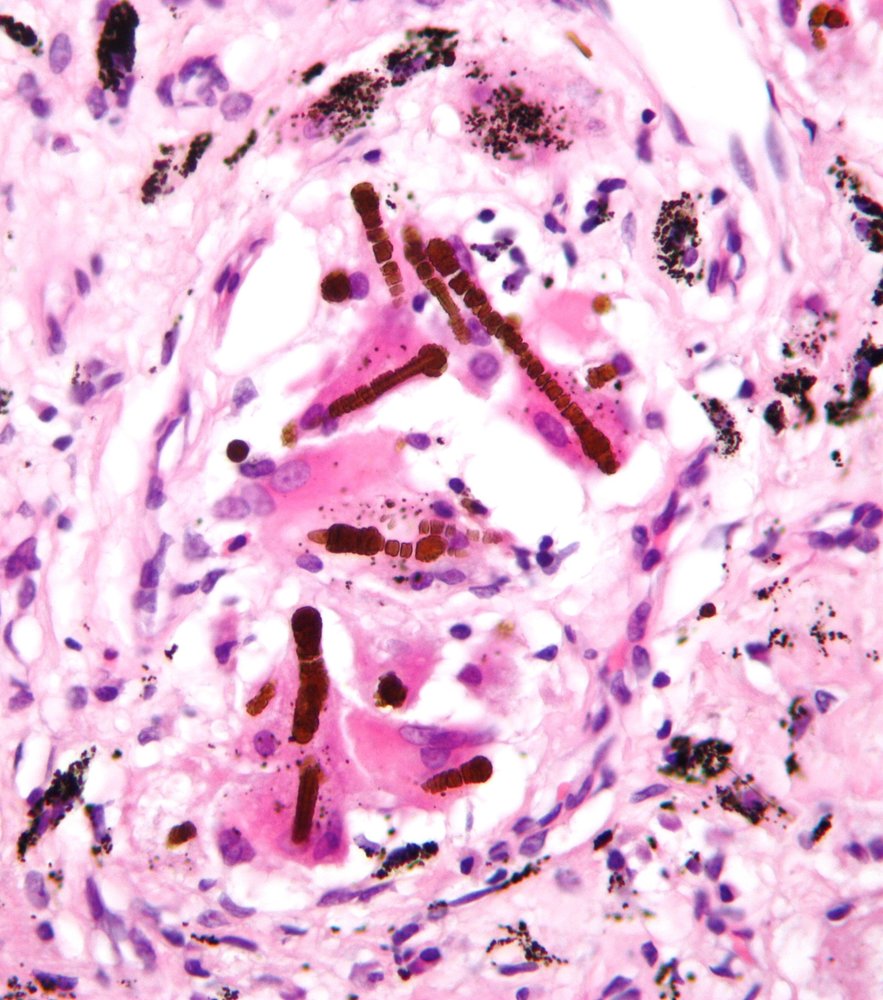
Photomicrograph of a lung biopsy (H&E stain; very high magnification)
There are multiple golden-brown, dumbbell-shaped ferruginous bodies (green overlay) adjacent to histiocytes and multinucleated giant cells within the fibrotic lung parenchyma. A transparent core is visible within some of the bodies. Clusters of anthracotic pigment (small black particles) are also visible.
The transparent core is characteristic of asbestos fibers. These findings are consistent with pulmonary asbestosis.
Source: “Ferruginous body” by Michael Bonert ("Nephron"), Wikimedia Commons, licensed under CC BY-SA 3.0. The supplementary image with overlays of relevant areas was adapted from the image mentioned above and licensed under CC BY-SA 3.0.
{kind=link}
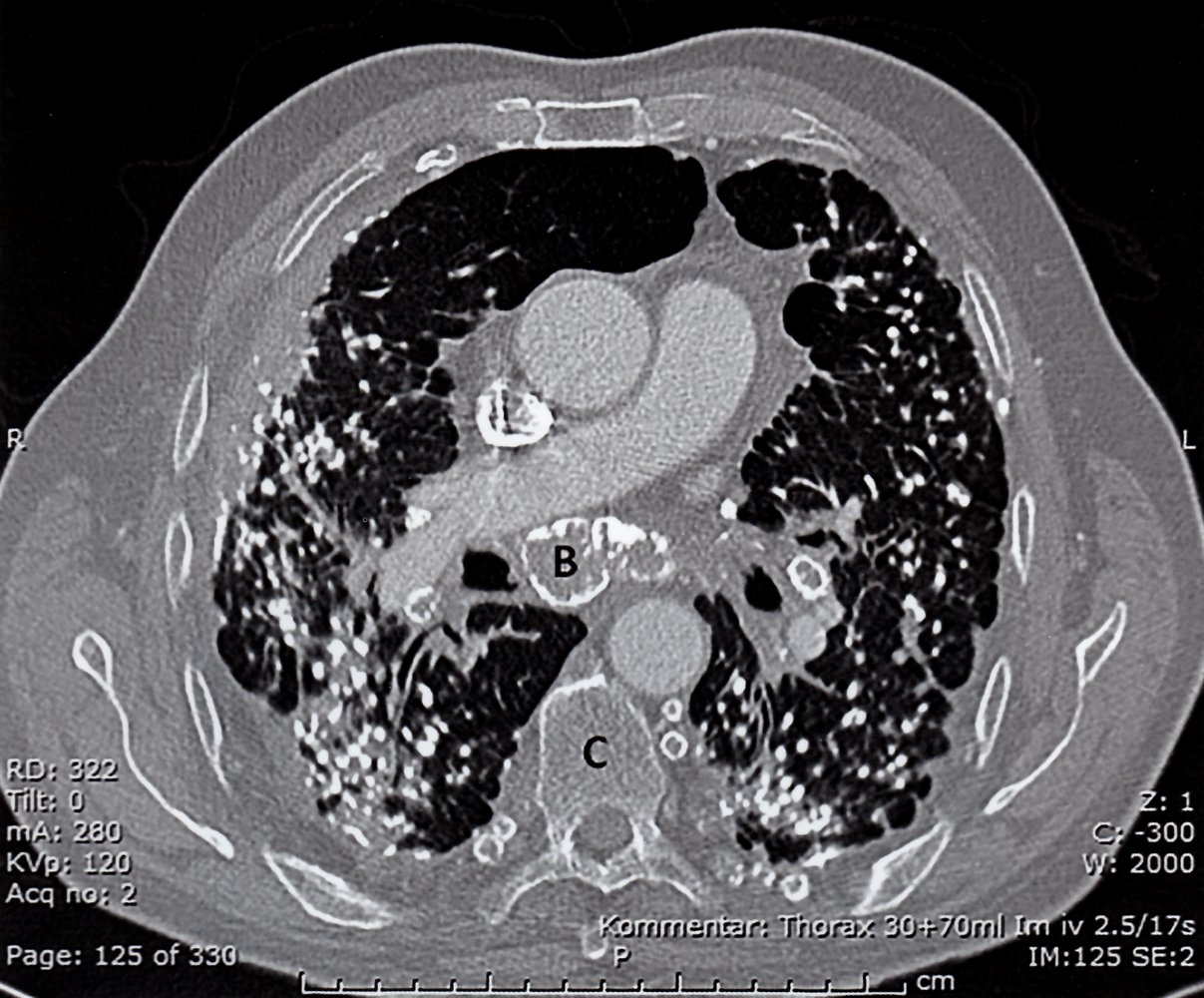
CT thorax (with contrast; axial plane) of a patient with history of exposure to silica dust
Innumerable small nodules are seen in both lungs, the majority of which are calcified (examples indicated by arrowheads). Some nodules have coalesced in the periphery of the lungs. Parenchymal opacities seen in association with nodules in the posterior lungs represent areas of developing progressive massive fibrosis (examples indicated by green hatched overlay). Peripheral calcification (eggshell calcification) is seen in several hilar and mediastinal lymph nodes (examples indicated by red outlines and B).
AA: ascending aorta; C: vertebral body; DA: descending aorta; P: right pulmonary artery; V: superior vena cava
Source: © IMPP
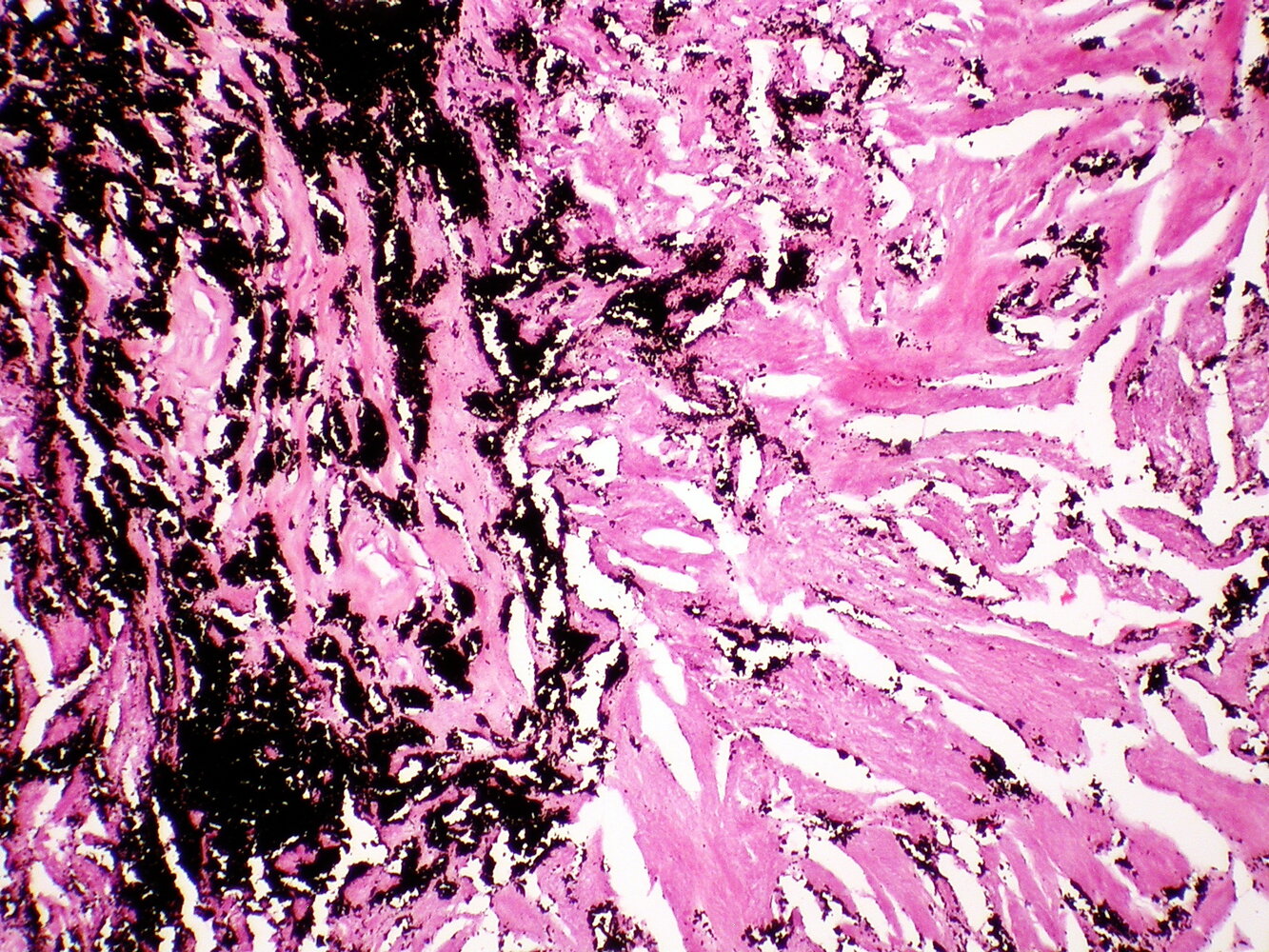
Photomicrograph of a lung biopsy specimen (H&E stain, low magnification)
Accumulations of carbon particles (anthracotic pigment) can be seen in the lung tissue. Some of the pigment can be found in pigment-laden macrophages. Also, collagenous fibrosis is predominant throughout the lung tissue.
This appearance is diagnostic of pneumoconiosis (e.g., coal workers' pneumoconiosis).
Source: “Coal workers pneumoconiosis - Anthracosilicosis” by Yale Rosen, Flickr, licensed under CC BY-SA 2.0.
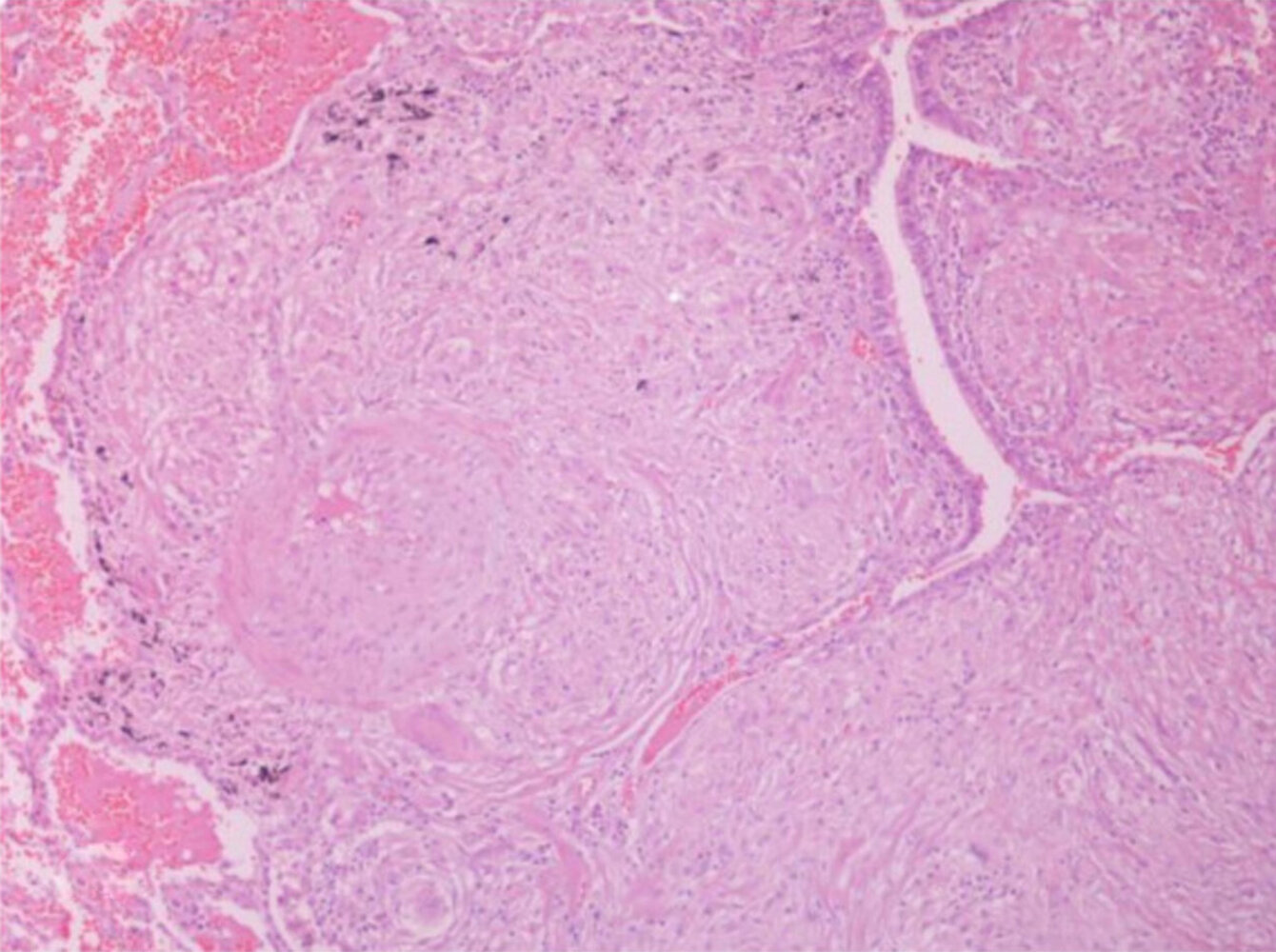
Photomicrograph of a lung biopsy specimen (H&E stain; 200x magnification)
Multiple confluent granulomas (green overlay) are visible within the bronchial submucosa, narrowing the lumen (arrow). Note the absence of central necrosis in the granulomas.
This appearance is characteristic of a noncaseating granuloma in sarcoidosis.
Source: “Figure 3, in: Sarcoidosis in a 65-year-old woman presenting with a lung mass and pericardial effusion: a case report” by G. A Margaritopoulos, A. Proklou, E. Lagoudaki et al., BMC - Journal of medical case reports, licensed under CC BY 2.0. Modifications: cropped white rim; removed letter "A". The supplementary image with overlays of relevant areas was adapted from the image mentioned above (© AMBOSS).
Pathophysiology
-
General
- All types of ILDs share the same basic pathophysiology.
- Repeated cycles of tissue injury in the lung parenchyma with aberrant wound healing → collagenous fibrosis → remodeling of the pulmonary interstitium [18]
- Pneumoconiosis: inhalation of dust particles →; phagocytosis by alveolar macrophages → destruction of alveolar macrophages, inflammatory reaction → scarring, granuloma formation
Clinical features
Features
-
Progressive dyspnea
- Exertional dyspnea that progresses to dyspnea at rest
- Patients may present with high-frequency, shallow breathing to compensate for dyspnea. [19]
- Persistent nonproductive cough
- Bibasilar, inspiratory crackles or rales (velcro-like rales) on auscultation
- Fatigue
- Fever is common in hypersensitivity pneumonitis and sarcoidosis, but otherwise uncommon.
- Findings suggestive of connective tissue disease, sarcoidosis, or vasculitis
Advanced disease
- Digital clubbing due to chronic hypoxia
- Cyanosis
- Loud inspiratory wheeze
Diagnosis
Approach [3][20][21]
- Perform a thorough clinical evaluation.
- Past medical history and family history for pulmonary and/or autoimmune conditions [3]
- Physical examination, including evaluation for signs of connective tissue disorders
- Obtain HRCT of the chest to evaluate for IPF and other alternative diagnoses. [22]
- Obtain laboratory studies to screen for CTD-ILD.
- Perform pulmonary function tests to assess disease severity and treatment response.
- If diagnosis remains unclear, consider:
- Additional laboratory studies based on clinical suspicion
- Invasive testing
Multidisciplinary care helps increase diagnostic accuracy. [3][23]
High-resolution CT (HRCT) chest [3][24][25]
Usual interstitial pneumonia (UIP) pattern is highly specific for IPF.
- Typical UIP pattern findings
- Honeycombing: multiple cystic lesions within the lung parenchyma due to fibrosis
- Irregular thickening of intralobular septa
- Reticular pattern and mild ground glass opacity (GGO)
- Traction bronchiectasis (irreversible dilatation of the bronchi and bronchioles due to fibrosis) or bronchiolectasis
- Pulmonary ossification may be seen.
- Other findings may be classified into:
- Probable UIP: some findings that support a diagnosis of IPF, e.g., a reticular pattern and traction bronchiectasis, but no honeycombing
- Indeterminate for UIP: nonspecific fibrosis that neither supports a diagnosis of IPF nor any alternative diagnosis
- Alternative diagnosis, e.g., hypersensitivity pneumonitis or sarcoidosis, can be made based on HRCT findings
- Interpretation
- If UIP pattern is present, a definitive diagnosis can be made without histopathologic confirmation.
- If other patterns are present, further evaluation is required to determine the cause.
In patients with IPF, the extent of abnormalities on HRCT correlates with the severity of functional impairment. [26]
")



")

Laboratory evaluation for CTD-ILD [3][23]
- Obtain in all patients:
-
Autoantibodies: ANA, rheumatoid factor and anti-CCP; , myositis-specific antibodies (e.g., anti-Jo-1 antibodies)
- May be elevated depending on underlying disease
- Consider additional studies on a case-by-case basis.
- Inflammatory markers: ↑ CRP, ↑ ESR
-
Autoantibodies: ANA, rheumatoid factor and anti-CCP; , myositis-specific antibodies (e.g., anti-Jo-1 antibodies)
- Obtain in patients with clinical features of myositis: muscle enzymes, e.g., ↑ aldolase
All patients should be screened for rheumatic and autoimmune diseases. [3][23]
Pulmonary function tests (PFTs)
PFTs can help assess disease severity but do not identify a specific cause of ILD. [22]
-
Restrictive lung disease pattern
- ↓ Total lung capacity and ↓ vital capacity
- Normal or ↓ FEV1
- ↓ FVC
- Normal or ↑ FEV1:FVC ratio
- Decreased diffusing capacity for CO (DLCO): highly sensitive parameter

Additional studies
X-ray chest [21]
Findings are not sufficient for a diagnosis but can raise clinical suspicion for ILD. [21]
- Normal in approx. 10% of patients
- Predominantly basilar increase in reticular opacities (sign of fibrosis)
- Patients may have nodular or mixed patterns.


Additional laboratory studies [21][22][24]
-
CBC with differential
- Anemia, e.g., anemia of chronic disease or due to occult pulmonary hemorrhage
- Eosinophilia, e.g., due to hypersensitivity pneumonitis or drug toxicity
- Liver enzymes: ↑ GGT, ↑ ALT, and/or ↑ AST may be consistent with sarcoidosis, amyloidosis, or polymyositis
- BMP: ↑ creatinine may be seen in CTD-ILD, pulmonary-renal syndromes, sarcoidosis, and amyloidosis
- Urinalysis: RBC casts, and/or dysmorphic RBCs may be seen in vasculitis and pulmonary-renal syndromes
-
ABG: nonspecific findings in patients with ILD
- ↑ Alveolar-arterial oxygen gradient
- ↓ PaO2
- Serum biomarkers for IPF: not recommended [3]
Invasive testing [21][27]
Obtain invasive testing if the diagnosis remains unclear and results will affect management.
- Bronchoscopy with bronchoalveolar lavage (BAL): Cellular analysis may help identify an underlying cause of ILD and exclude IPF. [3]
-
Surgical lung biopsy [21][28]
- Obtain if the diagnosis remains unclear after BAL.
- Biopsy findings may show a histopathologic pattern of definite, probable, or indeterminate UIP ; or an alternative diagnosis.
If HRCT shows a pattern of definite UIP, invasive testing is not usually necessary for diagnostic confirmation. [3]
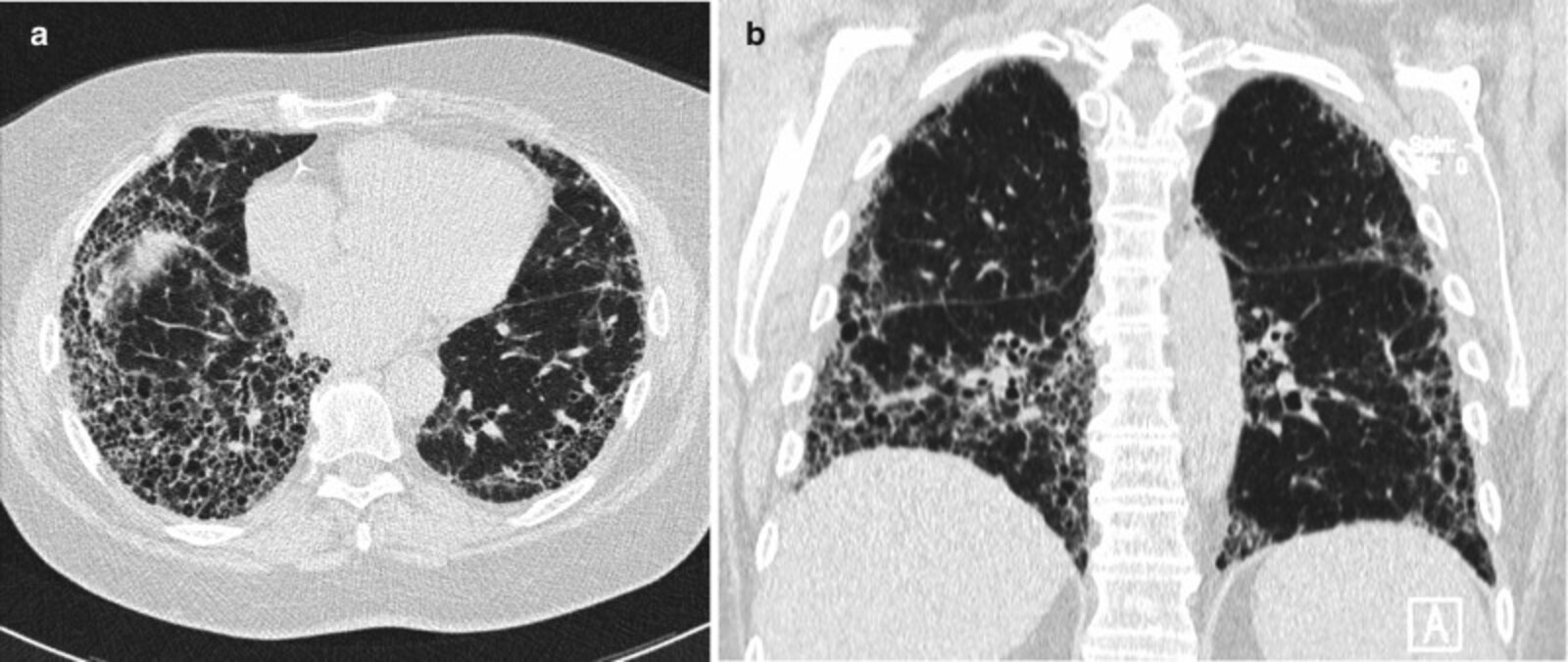
CT chest (lung window; a: axial plane; b: coronal plane) of a patient with progressive dyspnea on exertion
Extensive reticular interstitial thickening with honeycombing (examples indicated by green outlines) is accompanied by traction bronchiectasis (examples indicated by red arrows). The distribution is subpleural and basal predominant. The appearance is diagnostic of a usual interstitial pneumonia pattern. There are no CT features to suggest an alternative diagnosis.
DA: descending aorta; H: heart
Source: “Fig. 4.1, in: Chapter 4, Plain Film and HRCT Diagnosis of Interstitial Lung Disease” by Desai SR, Prosch H, Galvin JR, Diseases of the Chest, Breast, Heart and Vessels 2019-2022: Diagnostic and Interventional Imaging, licensed under CC BY 4.0. The supplementary image with overlays of relevant areas was adapted from the image mentioned above (© AMBOSS).
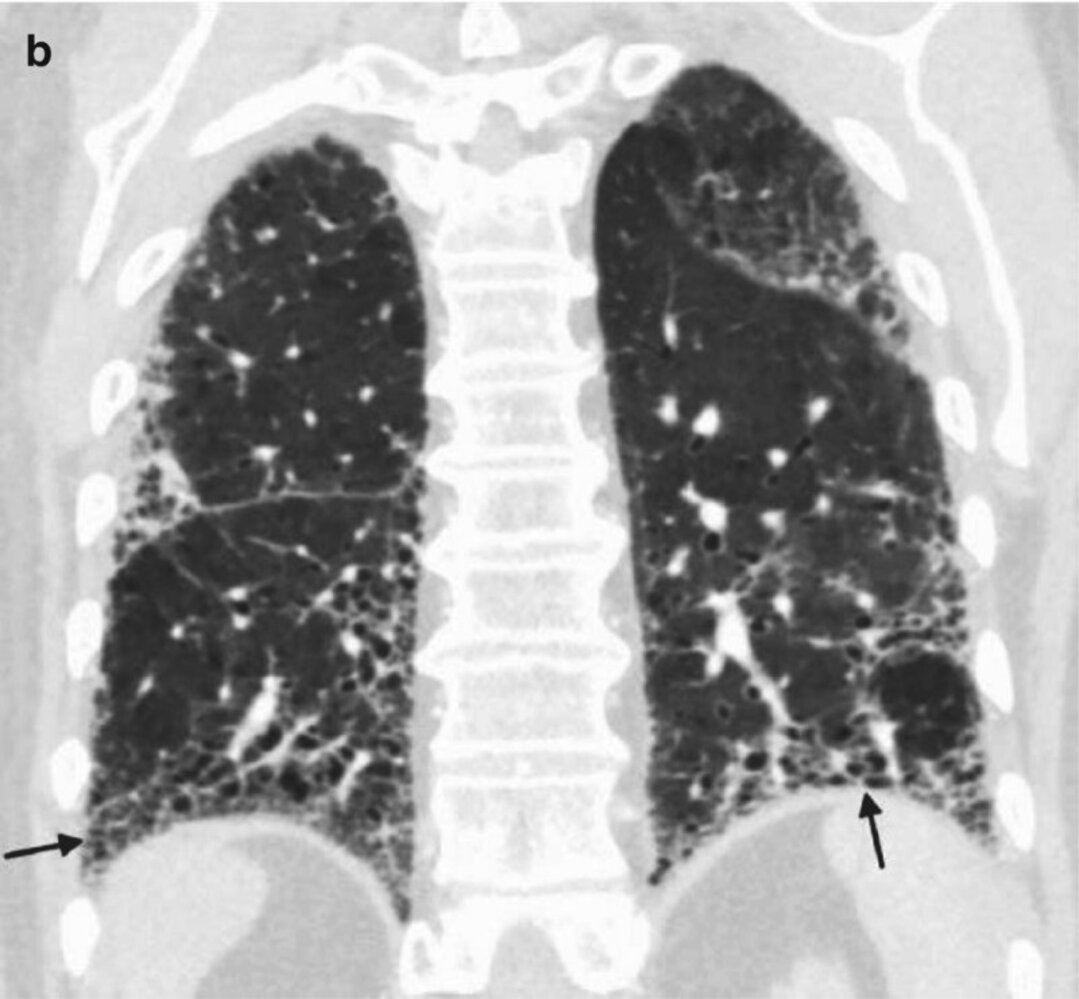
CT chest (coronal view; lung window) of a patient diagnosed with idiopathic pulmonary fibrosis
Coarse reticular opacities and clustered small cystic spaces (honeycomb lung; examples indicated by black arrows) with a basal predominant distribution are consistent with a usual interstitial pneumonia pattern. Traction bronchiectasis (examples indicated by white arrows) is visible.
Source: “Fig. 1.7b, in: Chapter 1, A Systematic Approach to Chest Radiographic Analysis” by Klein J, L. Rosado-de-Christenson, M, Diseases of the Chest, Breast, Heart and Vessels 2019-2022: Diagnostic and Interventional Imaging, licensed under CC BY 4.0. Modifications: image cropped. The supplementary image with overlays of relevant areas was adapted from the image mentioned above (© AMBOSS).
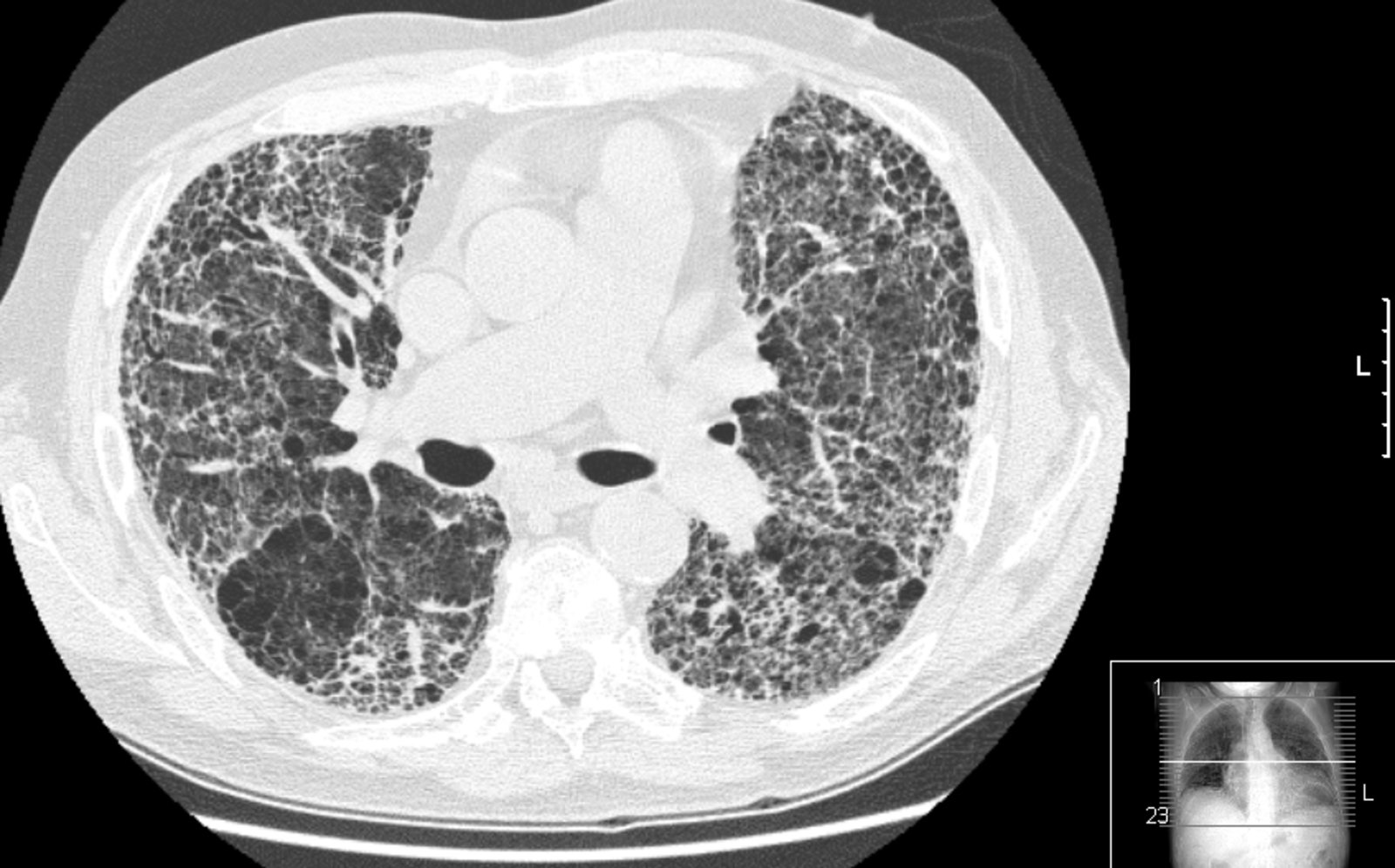
CT chest (high resolution; axial plane; lung window) of a patient with usual interstitial pneumonia
A diffuse reticular interstitial pattern is accompanied by traction bronchiectasis (examples indicated by green outline) and clustered small cystic air spaces (honeycombing). Multilayered honeycombing (example indicated by red outline) is conspicuous in the periphery of the lungs.
Source: “Pulmon fibrosis.PNG” by Drriad, Wikimedia Commons, licensed under CC BY-SA 3.0. The supplementary image with overlays of relevant areas was adapted from the image mentioned above and licensed under CC BY-SA 3.0.
{kind=link}
Click on the overlay icon below the taskbar on the left to toggle the overlay on and off.
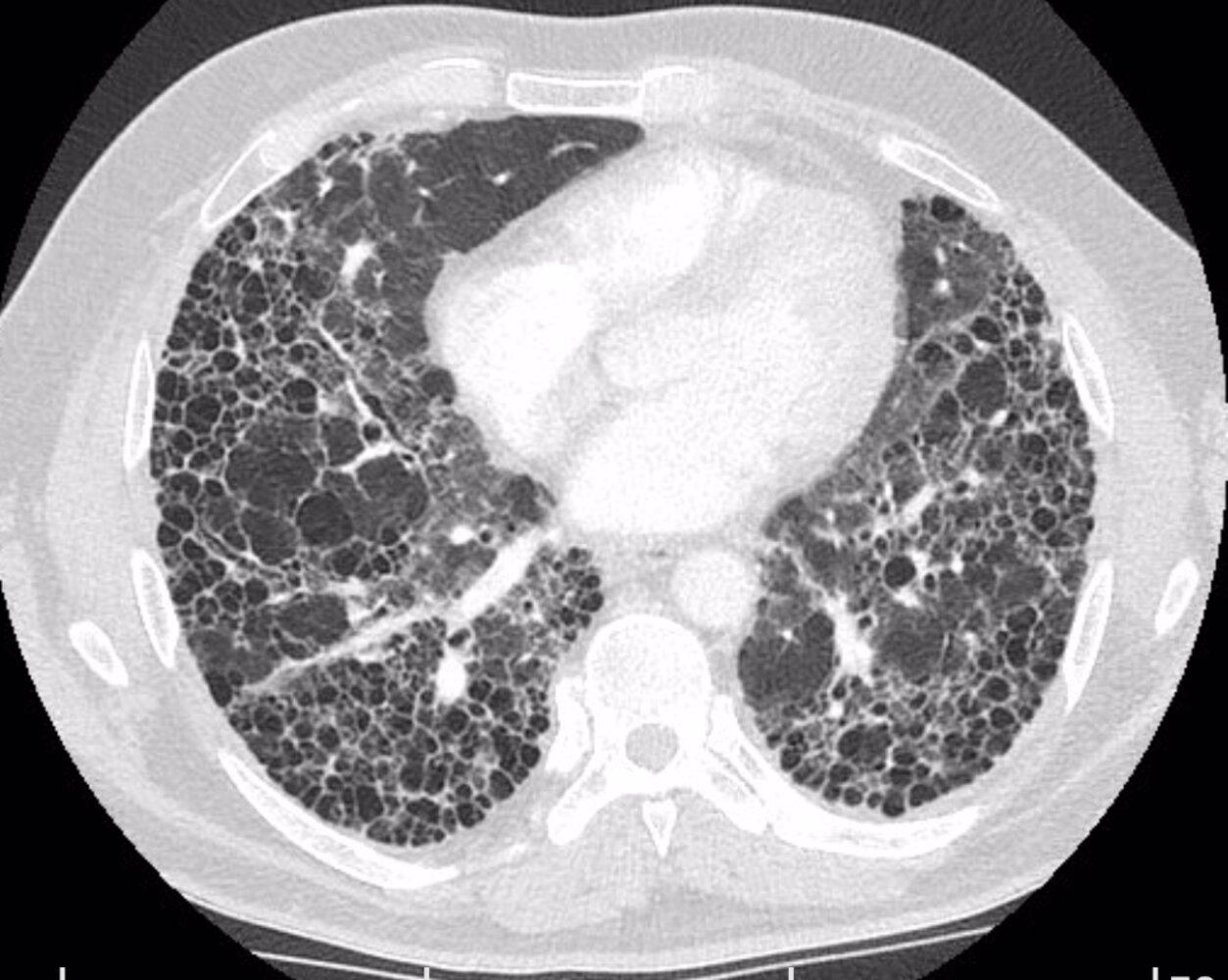
CT chest (with contrast; series 3: axial plane, mediastinal window; series 4: axial plane, lung window) of a patient with a history of work-related lignite and quartz dust exposure
There is evidence of chronic interstitial lung disease with extensive clustered cystic air space formation (honeycombing). Some areas show traction bronchiectasis. Enlarged reactive mediastinal and hilar lymph nodes are present. The main pulmonary artery is enlarged, which may indicate pulmonary hypertension. An indeterminate nodule is present in the right lung apex.
Image source of original image: Radiologie Frechen-Erftstadt. Original title: “Lung fibrosis”. Created by: Markus Le Blanc. Further notes: Many thanks to Dr. med. Markus Le Blanc (Radiologie Frechen-Erftstadt) for kindly granting permission for use of this image.
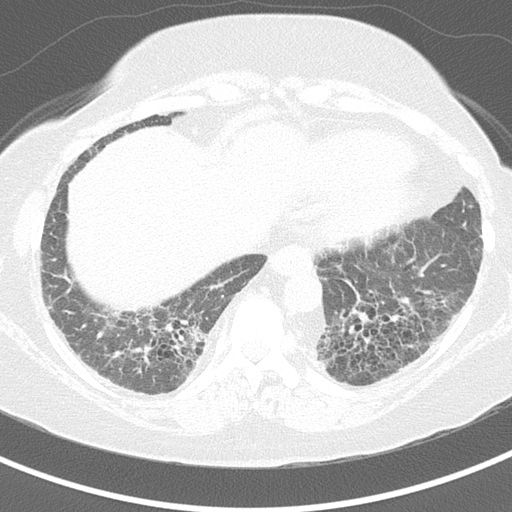
CT chest (high resolution; axial plane; lung window) of patient with chronic joint pain, dry cough, and shortness of breath
Extensive bibasal interstitial lung disease is present, with septal thickening, honeycombing, and traction bronchiectasis.Clinical and imaging findings suggest rheumatoid arthritis with rheumatoid-associated interstitial lung disease (RA-ILD).
Interstitial lung disease is one of the most common pulmonary manifestations of RA. The two main HRCT and histopathologic patterns in RA-ILD are usual interstitial pheumonia (UIP) and, less commonly, non specific interstitial pneumonia (NSIP).
© Massachusetts Medical Society. All rights reserved. AMBOSS SE, exclusive licensee.
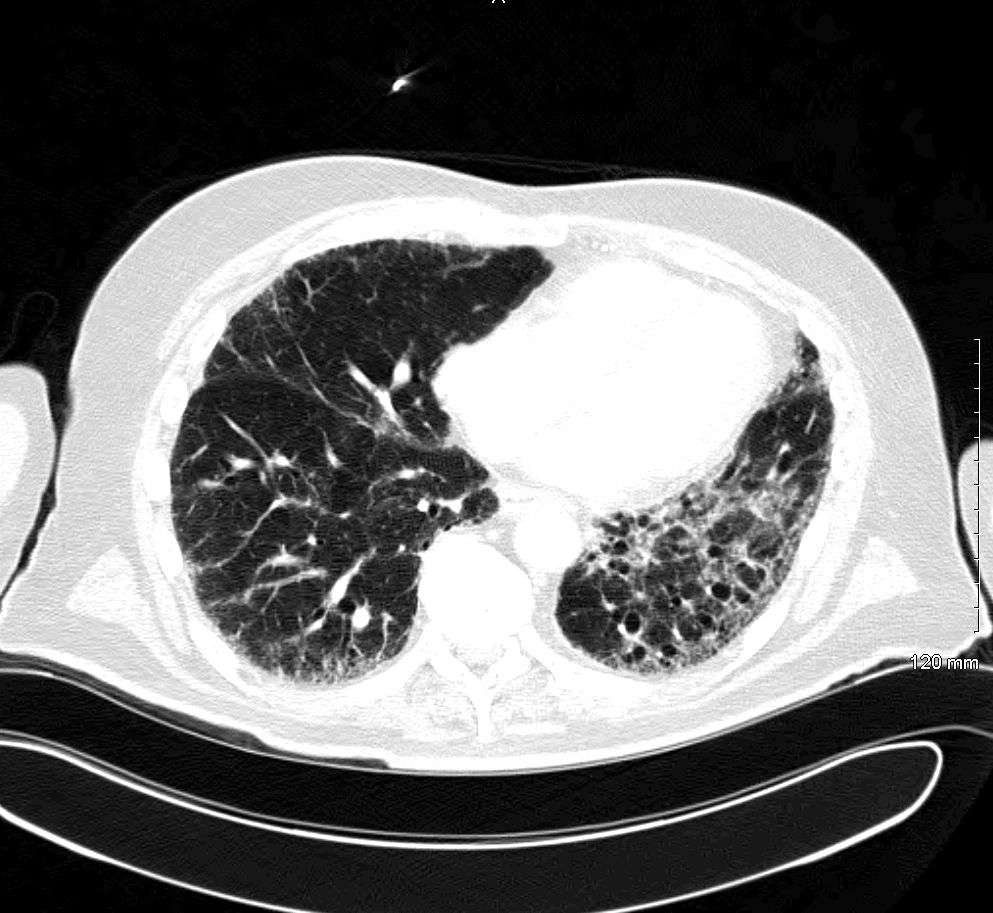
CT chest (axial plane; lung window) of middle-aged man with increased dyspnea on exertion
A bilateral lower lobe and right middle lobe reticular interstitial pattern is accompanied by mild ground glass (examples indicated by yellow overlay), bronchiectasis (examples indicated by arrows), and small cystic air spaces (honeycombing; examples indicated by blue overlay). There is volume loss in the left lung with ipsilateral mediastinal shift.
Biopsy showed usual interstitial pneumonia.
DA: descending aorta; H: heart
© Massachusetts Medical Society. All rights reserved. AMBOSS SE, exclusive licensee.
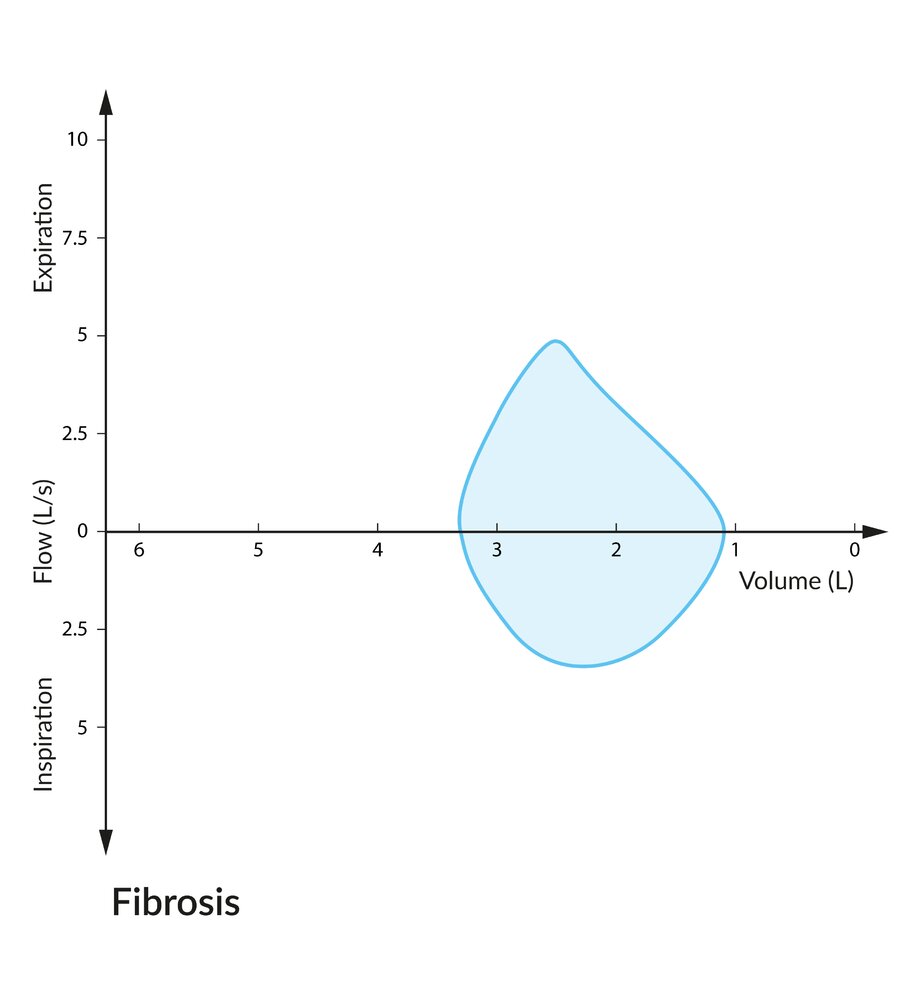
Total lung capacity (∼ 3.25 L) and peak expiratory and inspiratory flows are substantially decreased because of the lung's decreased compliance. Note that due to the enhanced elastic recoil of fibrosed lung parenchyma, expiratory flow is not decreased as greatly as lung volume.
© AMBOSS
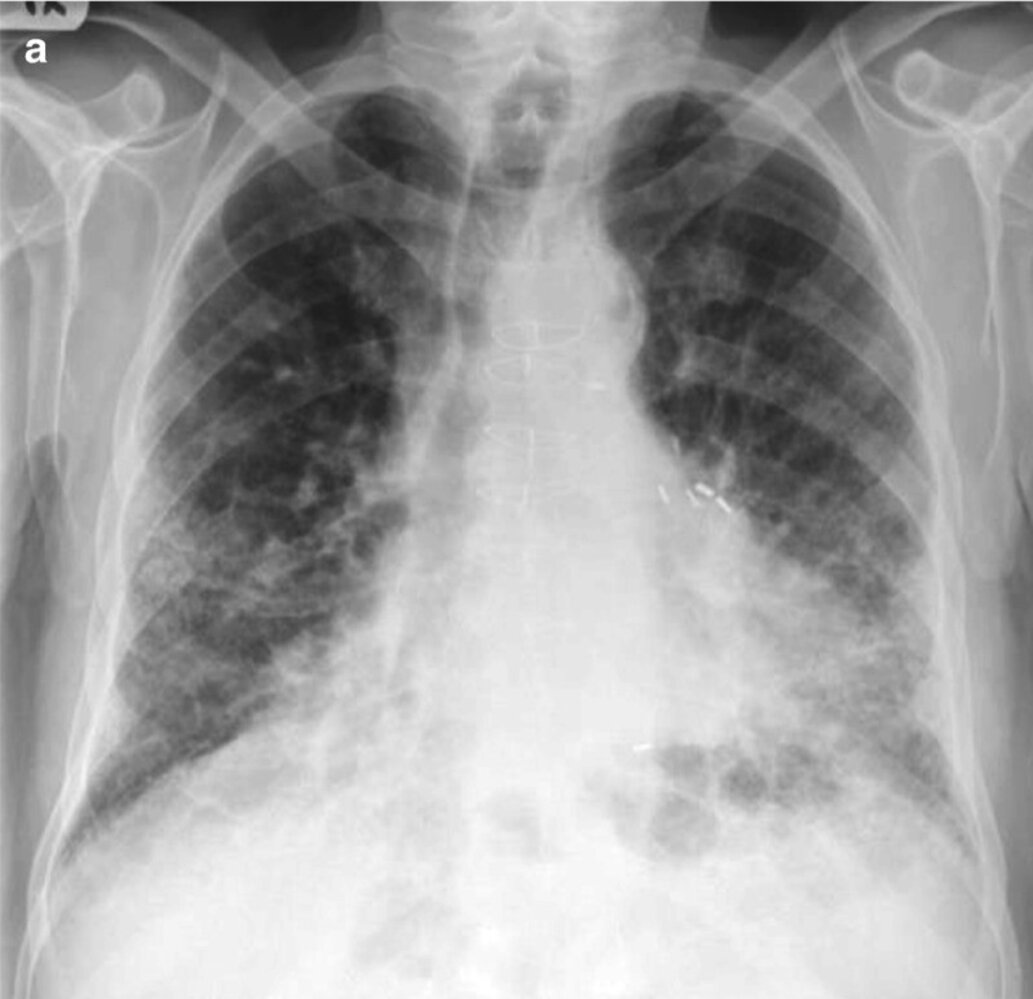
X-ray chest (posteroanterior view) of a patient with chronic progressive shortness of breath
Coarse reticular opacities (examples indicated by blue circles) in both lungs show a basal predominant distribution. The cardiac silhouette is enlarged and midline sternotomy wires (example of sternal wire indicated by black outline) and surgical clips (red lines) are evidence of prior coronary artery bypass grafting; however, there are no supportive findings for cardiogenic pulmonary edema, such as Kerley lines or pleural effusions.
CT was subsequently performed and showed peripheral honeycombing diagnostic of a usual-interstitial-pneumonia pattern. The patient was diagnosed with idiopathic pulmonary fibrosis (green overlay).
Source: “Fig. 1.7a, in: Chapter 1, A Systematic Approach to Chest Radiographic Analysis” by Klein J, L. Rosado-de-Christenson, M, Diseases of the Chest, Breast, Heart and Vessels 2019-2022: Diagnostic and Interventional Imaging, licensed under CC BY 4.0. Modifications: image cropped. The supplementary image with overlays of relevant areas was adapted from the image mentioned above (© AMBOSS).
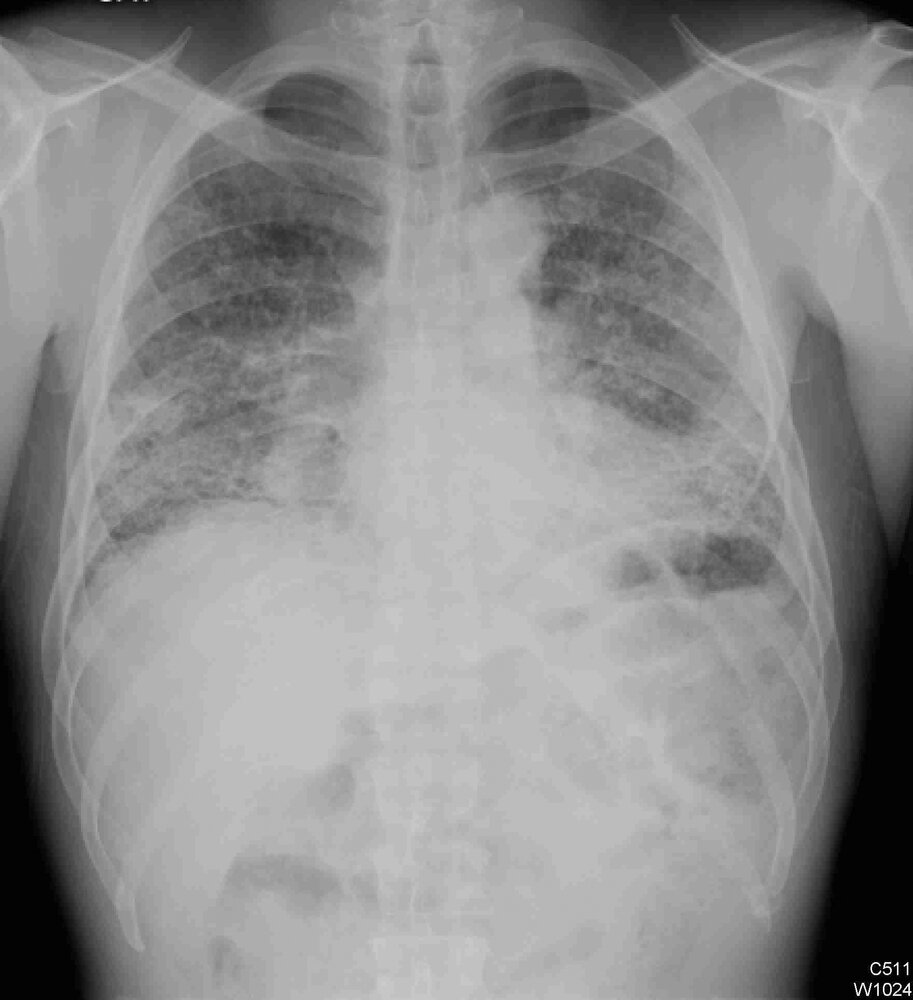
X-ray chest (PA view) of a patient with a history of idiopathic pulmonary fibrosis (IPF)
The lung volumes are low. Multilobar interstitial lung disease shows a lower zonal predominant distribution consistent with the clinical history of IPF.
© Massachusetts Medical Society. All rights reserved. AMBOSS SE, exclusive licensee.
Management
Approach [24][29]
- Offer supportive management, including preventive measures.
- Refer all patients to pulmonology for:
- Pharmacological therapy depending on the cause of ILD
- Evaluation for lung transplantation
- Identify and treat common comorbidities, e.g., GERD, pulmonary hypertension, major depressive disorder. [24][25][30]
- Initiate early advance care planning while patients are still able to participate in decision-making. [31]
ILD is a chronic, progressive, life-limiting condition. Encourage advance care planning and early engagement with palliative care. [29]
Supportive management [24]
- Encourage measures to prevent exacerbations and slow disease progression:
- Counsel on smoking cessation and treat tobacco-related disorders.
- Ensure recommended vaccinations are administered.
- Recommend avoidance of triggers for secondary causes of ILD.
- Offer pulmonary rehabilitation. [32][33]
- Supplementary oxygen therapy as needed
- Symptom management
- Consider cough suppressants and referral for multimodal speech therapy.
- Consider palliative pharmacotherapy for dyspnea as indicated, e.g., palliative anxiolysis. [31][34][35]
Treatment of IPF [24]
-
Pharmacological therapy
-
Antifibrotic agents may reduce mortality and acute exacerbations and slow the decline in FVC. [36][37][38]
- Pirfenidone: inhibits TGF-β-stimulated collagen synthesis
- Nintedanib: inhibits tyrosine kinases that target fibrogenic growth factors, e.g., VEGF, PDGF, and fibroblast growth factor receptor
- Immunosuppressive therapy is not indicated.
-
Antifibrotic agents may reduce mortality and acute exacerbations and slow the decline in FVC. [36][37][38]
- Lung transplantation: Refer for evaluation at the time of diagnosis. [39]
Do not start patients with IPF on immunosuppressive therapy; immunomodulation may worsen outcomes. [29]
Treatment of non-IPF ILD
-
Pharmacological therapy: depending on the cause of ILD [24]
- Treat the underlying condition if possible, e.g.:
- Specific management for patients with CTD-ILD
- Antibiotics if bacterial interstitial pneumonia is suspected
- Consider corticosteroids and/or systemic immunomodulators, e.g., for COP or NSIP, in consultation with a specialist.
- Treat the underlying condition if possible, e.g.:
-
Lung transplantation [39]
- In patients with fibrotic NSIP, refer for evaluation at the time of diagnosis.
- Refer patients with other types of non-IPF ILD and:
- Progression to respiratory failure
- ILD refractory to disease-specific therapies
Lung transplantation is the only curative option for ILD.
Management of acute exacerbation of IPF (AE-IPF) [40][41]
AE-IPF is characterized by acute respiratory deterioration accompanied by new widespread alveolar abnormalities and it may be idiopathic or triggered. [41]
- Clinical features may include: [40]
- Worsening cough with increased sputum production
- Fever
- Hypoxemia
- Diagnose AE-IPF if all four clinical diagnostic criteria are present : [41]
- Known diagnosis of IPF
- Worsening dyspnea for < 1 month
- New bilateral ground-glass opacities and/or consolidation over a background of UIP on HRCT chest
- Symptoms are not explained by an alternative diagnosis.
- Management [40][41]
- Admit to hospital.
- Consider ICU admission and mechanical ventilation to bridge eligible patients to lung transplantation.
- Offer supportive care, e.g., supplemental oxygen, palliative treatment for dyspnea.
- High-dose systemic corticosteroids and/or antibiotic therapy may be considered.
- Consult the patient's pulmonologist as soon as possible.
- Discuss goals of care and consult palliative care if indicated.
AE-IPF has an in-hospital mortality rate of ∼ 50%. An elevated APACHE score, the need for mechanical ventilation, and hypoxemia are all associated with increased mortality in hospitalized patients with ILD. [42]
Complications
- Pulmonary hypertension
- Cor pulmonale
- Respiratory failure: initially partial respiratory failure (↓ pO2), followed by global respiratory failure (↑ pCO2) in advanced stages
We list the most important complications. The selection is not exhaustive.
External Resources
References
- American Thoracic Society. "American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias". Am J Respir Crit Care Med. 165(2). :277-304. (2002)
- Thomas Brack, Amal Jubran, Martin J. Tobin. "Dyspnea and Decreased Variability of Breathing in Patients with Restrictive Lung Disease". Am J Respir Crit Care Med. 165(9). :1260-1264. (2002)
- Raghu G, Remy-Jardin M, Myers JL, et al. "Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline". Am J Respir Crit Care Med. 198(5). :e44-e68. (2018)
- Wong AW, Fidler L, Marcoux V, et al. "Practical Considerations for the Diagnosis and Treatment of Fibrotic Interstitial Lung Disease During the Coronavirus Disease 2019 Pandemic". Chest. 158(3). :1069-1078. (2020)
- Mikolasch TA, Garthwaite HS, Porter JC. "Update in diagnosis and management of interstitial lung disease". Clin Med (Northfield Il). 17(2). :146-153. (2017)
- Kalchiem-Dekel O, Galvin J, Burke A, Atamas S, Todd N. "Interstitial Lung Disease and Pulmonary Fibrosis: A Practical Approach for General Medicine Physicians with Focus on the Medical History". J Clin Med. 7(12). :476. (2018)
- Furini F, Carnevale A, Casoni GL, et al. "The Role of the Multidisciplinary Evaluation of Interstitial Lung Diseases: Systematic Literature Review of the Current Evidence and Future Perspectives". Front. Med. 6. (2019)
- Meyer KC. "Diagnosis and management of interstitial lung disease". Transl Respir Med. 2(1). (2014)
- Raghu G, Remy-Jardin M, Richeldi L, et al. "Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline". Am J Respir Crit Care Med. 205(9). :e18-e47. (2022)
- Nyquist A, Mushtaq R, Gill F, Yaddanapudi K. "High-Resolution Computed Tomography Evaluation of Interstitial Lung Disease for the Pulmonologist". Curr Pulmonol Rep. 9(4). :119-130. (2020)
- Kebbe J, Abdo T. "Interstitial lung disease: the diagnostic role of bronchoscopy". J Thorac Dis. 9(S10). :S996-S1010. (2017)
- Kaarteenaho R. "The current position of surgical lung biopsy in the diagnosis of idiopathic pulmonary fibrosis". Respir Res. 14(1). :43. (2013)
- Karkhanis VS, Joshi JM. "Pneumoconioses". Indian J Chest Dis Allied Sci. 55(1). :25-34. (2013)
- Kim K-I, Kim CW, Lee MK, et al. "Imaging of Occupational Lung Disease". RadioGraphics. 21(6). :1371-1391. (2001)
- O'Reilly KM, Mclaughlin AM, Beckett WS, Sime PJ. "Asbestos-related lung disease". Am Fam Physician. 75(5). :683-8. (2007)
- Mir WAY, Poudel A, Adhikari A, et al. "Asbestos-Related Diseases and Its Impact on Health: An Updated Review Article". Curr Pulmonol Rep. 12(4). :244-255. (2023)
- Smolkova P, Nakladalova M. "The etiology of occupational pulmonary aluminosis - the past and the present". Biomedical Papers. 158(4). :535-538. (2014)
- Smolková P, Nakládalová M, Tichý T, Hampalová M, Kolek V. "Occupational pulmonary aluminosis: a case report". Ind Health. 52(2). :147-151. (2014)
- Leonard R, Zulfikar R, Stansbury R. "Coal mining and lung disease in the 21st century". Curr Opin Pulm Med. 26(2). :135-141. (2020)
- Mirsadraee M. "Anthracosis of the lungs: etiology, clinical manifestations and diagnosis: a review". Tanaffos. 13(4). :1-13. (2014)
- Dweik RA. "Berylliosis Medication". WebMD. https://emedicine.medscape.com/article/296759-medication. [2015-12-31]
- Boffetta P, Fordyce TA, Mandel JS. "A mortality study of beryllium workers". Cancer Medicine. 5(12). :3596-3605. (2016)
- Khalid I, Khalid TJ, Jennings JH. "A welder with pneumosiderosis: a case report". Cases Journal. 2(1). :6639. (2009)
- Akar E, Yildiz T, Atahan S. "Pulmonary siderosis cases diagnosed with minimally invasive surgical technique: A retrospective analysis of 7 cases". Annals of Thoracic Medicine. 13(3). :163. (2018)
- Patel PH, Yarrarapu SNS, Anjum F. "Byssinosis". Statpearls. (2021)
- Sauleda J, Núñez B, Sala E, Soriano J. "Idiopathic Pulmonary Fibrosis: Epidemiology, Natural History, Phenotypes". Medical Sciences. 6(4). :110. (2018)
- Chandra D, Maini R, Hershberger DM. "Cryptogenic Organizing Pneumonia". StatPearls. (2021)
- Raghu G, Rochwerg B, Zhang Y, et al. "An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline". Am J Respir Crit Care Med. 192(2). :e3-e19. (2015)
- Nathan SD, Barbera JA, Gaine SP, et al. "Pulmonary hypertension in chronic lung disease and hypoxia". Eur Respir J. 53(1). :1801914. (2019)
- Gersten RA, Moale AC, Seth B, et al. "A scoping review of palliative care outcome measures in interstitial lung disease". Europ Respir Rev. 30(161). :210080. (2021)
- Bell EC, Cox NS, Goh N, et al. "Oxygen therapy for interstitial lung disease: a systematic review". Eur Respir Rev. 26(143). :160080. (2017)
- Dowman L, Hill CJ, May A, Holland AE. "Pulmonary rehabilitation for interstitial lung disease". Cochrane Database of Systematic Reviews. 2021(2). (2021)
- Smallwood N, Thompson M, Warrender-Sparkes M, et al. "Integrated respiratory and palliative care may improve outcomes in advanced lung disease". ERJ Open Res. 4(1). :00102-2017. (2018)
- Jabbarian LJ, Zwakman M, van der Heide A, et al. "Advance care planning for patients with chronic respiratory diseases: a systematic review of preferences and practices". Thorax. 73(3). :222-230. (2017)
- Meyer K, Decker C. "Role of pirfenidone in the management of pulmonary fibrosis". Ther Clin Risk Manag. 13. :427-437. (2017)
- Margaritopoulos G, Vasarmidi E, Antoniou K. "Pirfenidone in the treatment of idiopathic pulmonary fibrosis: an evidence-based review of its place in therapy". Core Evid. 11. :11-22. (2016)
- Knüppel L, Ishikawa Y, Aichler M, et al. "A Novel Antifibrotic Mechanism of Nintedanib and Pirfenidone. Inhibition of Collagen Fibril Assembly". Am J Respir Cell Mol Biol. 57(1). :77-90. (2017)
- Kapnadak SG, Raghu G. "Lung transplantation for interstitial lung disease". Eur Respir Rev. 30(161). :210017. (2021)
- Leuschner G, Behr J. "Acute Exacerbation in Interstitial Lung Disease". Front Med. 4. (2017)
- Collard HR, Ryerson CJ, Corte TJ, et al. "Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report". Am J Respir Crit Care Med. 194(3). :265-275. (2016)
- Huapaya JA, Wilfong EM, Harden CT, Brower RG, Danoff SK. "Risk factors for mortality and mortality rates in interstitial lung disease patients in the intensive care unit". European Respiratory Review. 27(150). :180061. (2018)
- Glasser SW, Hardie WD, Hagood JS. "Pathogenesis of Interstitial Lung Disease in Children and Adults". Pediatric Allergy, Immunology, and Pulmonology. 23(1). :9-14. (2010)