Summary
Multiple endocrine neoplasia (MEN) is a term used to describe three autosomal dominant syndromes that are associated with certain hormone-producing neoplasias. There are three subtypes: MEN 1, MEN 2A, and MEN 2B. MEN 1 is caused by an altered menin protein expression and presents with primary hyperparathyroidism, often in association with endocrine pancreatic tumors and/or pituitary adenomas. MEN 2A and MEN 2B are caused by a mutated RET proto-oncogene and both present with medullary thyroid carcinoma and sometimes pheochromocytoma. MEN 2A is further associated with primary hyperparathyroidism as well, while MEN 2B causes a marfanoid habitus and sometimes neurinomas. If any of the individual conditions associated with MEN are suspected, especially in patients with a positive family history, it is important to consider a diagnostic workup for any of the other associations. Specific diagnostic and management approaches can be found within the articles for each of the individual conditions. Family members of MEN patients should receive genetic counseling. Those positive for mutated genes should be closely monitored and should undergo a total thyroidectomy if positive for the RET proto-oncogene.
Overview
| Overview of multiple endocrine neoplasia | |||
|---|---|---|---|
| Feature | MEN 1 (formerly known as Wermer syndrome) | MEN 2 | |
| MEN 2A (formerly known as Sipple syndrome) | MEN 2B | ||
| Genetics |
|
|
|
| Inheritance |
|
||
| Principal manifestations |
|
|
|
| Further manifestations |
|
|
|
|
|
||
| Management |
|
|
|
MEN 1: 3 "P"s = Parathyroid, Pancreas, Pituitary gland
MEN 2A: 1 "M", 2 "P”s = Medullary thyroid carcinoma, Pheochromocytoma, Parathyroid
MEN 2B: 2 “M”s, 1 “P” = Medullary thyroid carcinoma, Marfanoid habitus/Multiple neuromas, Pheochromocytoma
Remembering that the inheritance pattern of MEN syndromes is autosomal dominant requires MENtal dominance.
 syndromes")

References:[1][2][3][4]
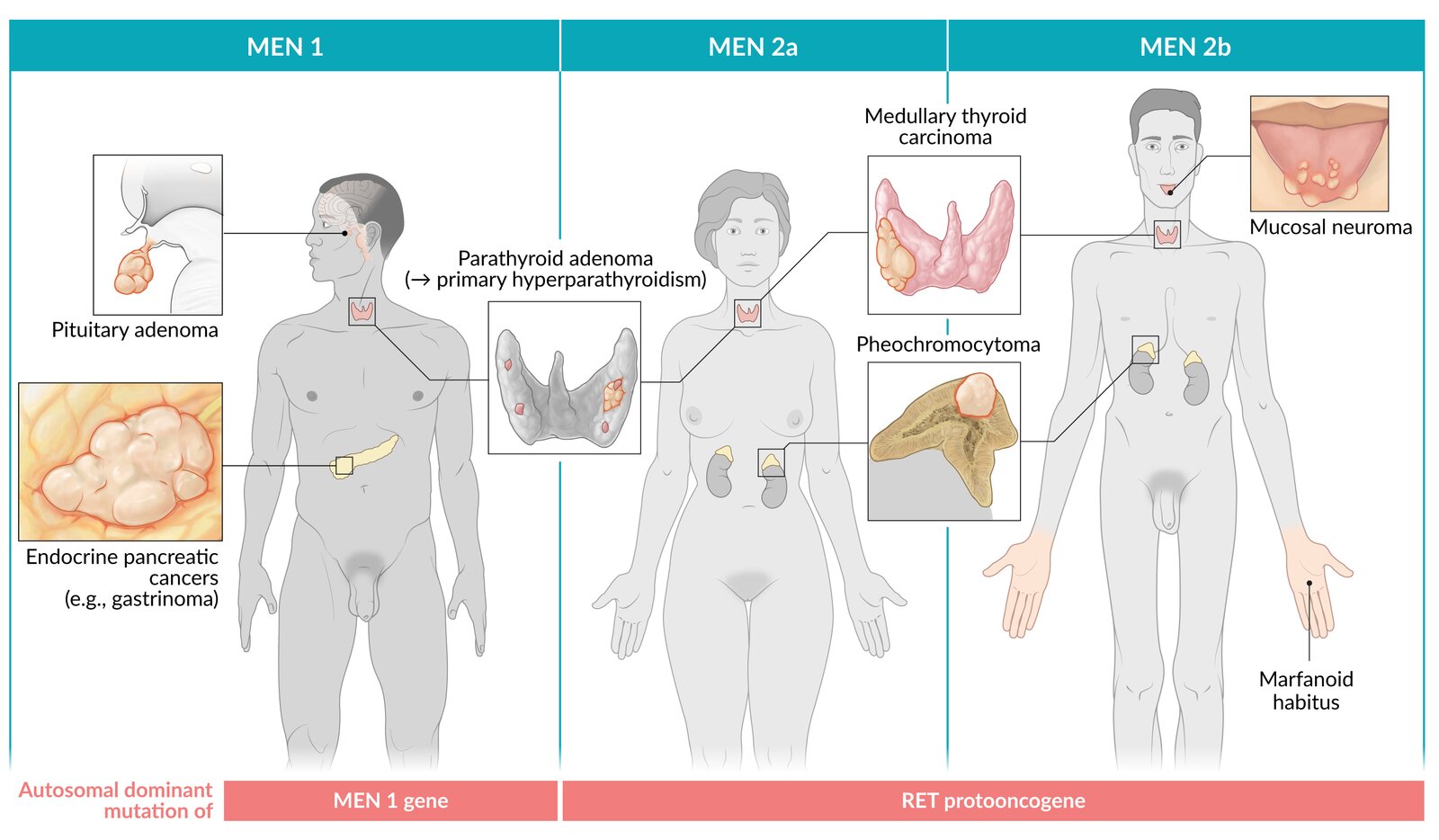
© AMBOSS
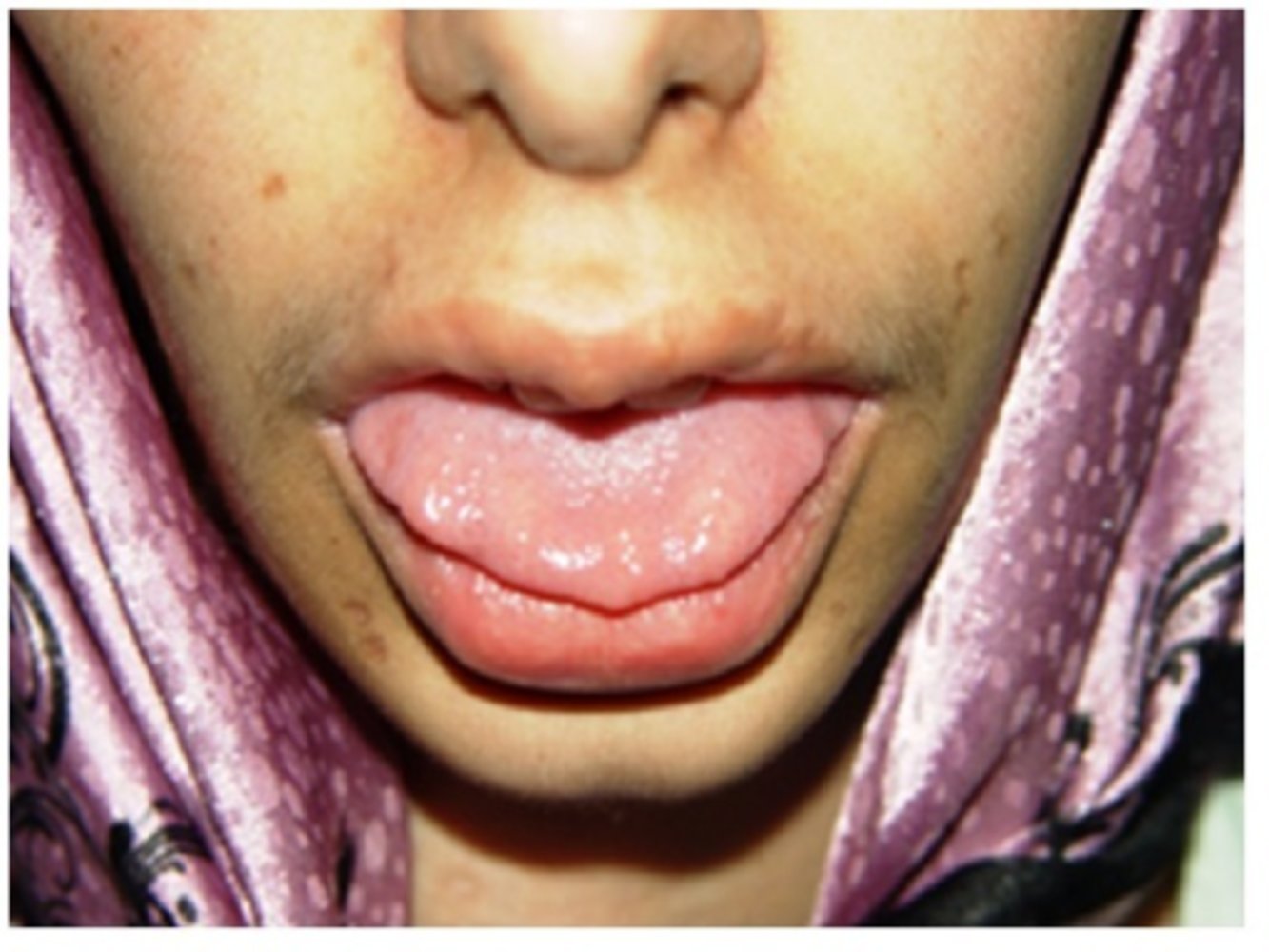
Photograph of the tongue and lips of an 18-year-old woman with multiple endocrine neoplasia type 2b
Multiple raised subcutaneous nodules are seen at the edge of the tongue and upper lip.
These nodules are characteristic of mucosal neuroma.
Source: “Figure 1. in: A Case of Multiple Endocrine Neoplasia Type 2B and Gangliomatosis of Gastrointestinal Tract” by Banafshe Shahnazari, Aria Aghamaleki, Bagher Larijani, Mohammad Reza Mohajeri Tehrani, Hasan Rafati, and Abdolreza Babamahmoodi, Case Reports in Medicine, Hindawi, licensed under CC BY-SA 3.0.
References
- "Pheochromocytomas in Multiple Endocrine Neoplasia Type 2". https://www.ncbi.nlm.nih.gov/pubmed/26494388?dopt=Abstract. [2015-01-01]
- Alevizaki M, Saltiki K. "Primary Hyperparathyroidism in MEN2 Syndromes". Recent Results Cancer Res. 204. :179-186. (2015)
- Editors: Warrell DA, Cox TM, Firth JD; Contributor: Benz, Jr EJ. "Oxford Textbook of Medicine". Oxford University Press. 1(4). (2003). ISBN: 019852787x
- Kasper DL, Fauci AS, Hauser SL, et al. "Harrison's Principles of Internal Medicine". McGraw-Hill Education. (2015). ISBN: 9780071802161
- Williams L. "Wermer Syndrome (MEN Type 1)". WebMD. http://emedicine.medscape.com/article/126438-overview#showall. [2015-02-18]
- Arnold A. "Multiple endocrine neoplasia type 1: Clinical manifestations and diagnosis". UpToDate. UpToDate. https://www.uptodate.com/contents/multiple-endocrine-neoplasia-type-1-clinical-manifestations-and-diagnosis#H28380297. [2015-05-28]
- Author: Melanie L Richards. "Type 2 Multiple Endocrine Neoplasia". WebMD. http://emedicine.medscape.com/article/123447-overview#showall. [2015-12-11]