Summary
Myeloproliferative neoplasms (MPNs) are a group of disorders characterized by a proliferation of normally developed (nondysplastic) multipotent hematopoietic stem cells from the myeloid cell line. The most common (classic) MPNs are chronic myeloid leukemia (CML), essential thrombocythemia (ET), polycythemia vera (PV), and primary myelofibrosis (PMF). Clinical findings overlap significantly between these conditions and the initial diagnostic workup is the same, including blood work (e.g., CBC, peripheral smear), genetic testing, and, if needed, bone marrow studies. Blood tests demonstrate myeloid cell proliferation: Granulocytes are increased in CML, thrombocytes in ET, and all three myeloid lines are increased in PV. PMF has an initial hyperproliferative phase often followed by pancytopenia as a result of progressive bone marrow failure. Elevated uric acid levels and gout may be seen in all MPNs as a result of increased cellular breakdown. Genetic markers can provide further information for diagnosis and also help guide treatment. The Philadelphia chromosome is present in almost all cases of CML. Driver mutations (e.g., JAK2, CALR, MPL) affecting the JAK-STAT signaling pathway are the main diagnostic markers for the remaining classic MPNs. Less common MPNs, which are not associated with the driver mutations, include chronic eosinophilic leukemia (CEL), chronic neutrophilic leukemia, and myeloproliferative neoplasm, unclassifiable. Treatments for all MPNs primarily focus on the prevention of known complications (e.g., thrombohemorrhagic events) and the alleviation of symptoms with combinations of medications and/or procedures including platelet inhibitors, cytoreduction, phlebotomy, targeted therapy, transfusions of blood products, and splenectomy. Patients are kept under close observation because of the risk of developing acute myeloid leukemia (AML). Allogeneic hematopoietic stem cell transplantation is the only curative treatment.
See also ”Polycythemia vera” and ”Chronic myeloid leukemia” for further detail on these conditions.
Overview of myeloproliferative neoplasms
Conditions [1]
According to the WHO classification, the following disorders are myeloproliferative neoplasms:
-
Common (classic)
- Essential thrombocythemia
- Polycythemia vera
- Primary myelofibrosis
- Chronic myeloid leukemia
-
Less common
- Chronic eosinophilic leukemia
- Chronic neutrophilic leukemia
- Myeloproliferative neoplasms, unclassifiable
All MPNs can potentially progress to AML. [2]






Clinical features [3]
These can occur with varying frequency, depending on the underlying MPN.
- Constitutional symptoms, especially fatigue
- Abdominal pain
- Hepatosplenomegaly
- Thrombohemorrhagic events
- Features of hyperuricemia, e.g., gouty arthritis
Diagnostics [1][4]
- Indications include:
- Characteristic clinical symptoms
- Abnormal physical examination findings (e.g., hepatosplenomegaly)
- MPNs may also be seen incidentally when routine blood work shows abnormal cell counts on CBC.
Initial studies
- CBC: detects elevated or decreased myeloid cell lines
- Peripheral smear: detects morphological changes in myeloid cell lines
- Elevated LDH, uric acid, and/or leukocyte alkaline phosphatase: indicates high disease burden and high cell turnover
- CT or ultrasound abdomen: Hepatosplenomegaly may be present.

Confirmatory studies [5][6][7]
-
Genetic testing: required to diagnose MPNs
- Initial mutation screening
- Philadelphia chromosome (BCR-ABL): present in almost all cases of CML
- Driver mutations (e.g., JAK2, CALR, MPL): associated with PV, ET, and PMF
- Subsequent mutation testing: Extended panels may detect atypical driver mutations, nondriver mutations (e.g., CSF3R), and high-molecular risk mutations. [7]
- Initial mutation screening
- Bone marrow biopsy: often required to definitively confirm a diagnosis
With the exception of CML, all of the classic MPNs have varying degrees of JAK2 mutations, which can be used as a diagnostic marker. [7][8]
Diagnostic comparison of classic MPNs [1][4]
The WHO has established diagnostic criteria for each of the MPNs (See “Tips and links” for full criteria). Laboratory findings seen in classic MPNs are included in the following table.
| Comparison of classic myeloproliferative neoplasms [1][4][6] | |||
|---|---|---|---|
| Clinical and laboratory studies | Percentage of patients with mutations [7][8] | Bone marrow | |
| Essential thrombocythemia |
|
|
|
| Polycythemia vera |
|
|
|
| Primary myelofibrosis |
|
|
|
| Chronic myeloid leukemia |
|
|
|
MPNs have overlapping presenting features, mutations, and diagnostic findings. A patient's diagnosis may change if their condition progresses over time (e.g., PV may transform into myelofibrosis; PMF may transform into AML). [4][11]
Treatment overview [8][12]
The choice of treatment depends on the underlying diagnosis, the presence of risk factors, and any comorbidities. [8]
- Low-risk MPNs are often managed with observation.
- High-risk or symptomatic MPNs are managed with a combination of symptomatic management, treatment to delay disease progression, and, in rare cases, allogeneic stem cell transplantation.
Supportive care
-
Thromboembolism prevention
- Antiplatelet drugs (e.g., aspirin)
- Therapeutic phlebotomy: to decrease hematocrit and blood viscosity for high RBC counts
-
Symptomatic splenomegaly [13]
- Splenectomy, splenic irradiation
- JAK2 inhibitors
- Cytoreduction: Options include hydroxyurea, cladribine, and interferon alpha. [8]
-
Symptomatic cytopenias (e.g., bleeding from thrombocytopenia, symptomatic anemia)
- Transfusions (e.g., blood, platelets)
- Erythropoietin and its analogs (e.g., darbepoetin)
- Androgenic steroids (e.g., danazol) [14][15]
- Prednisone
- Immunomodulators: thalidomide and analogs (e.g., lenalidomide, pomalidomide) [14]
- Splenectomy for refractory transfusion-dependent anemia
-
Relief of associated symptoms
- Gouty arthritis: allopurinol
- Pruritus: antihistamines
Disease-specific therapy
- Targeted therapy: JAK2 inhibitors (e.g., ruxolitinib) for PV and myelofibrosis [8]
- Allogeneic hematopoietic stem cell transplantation: potentially curative [8]

© AMBOSS

© AMBOSS

© AMBOSS

© AMBOSS
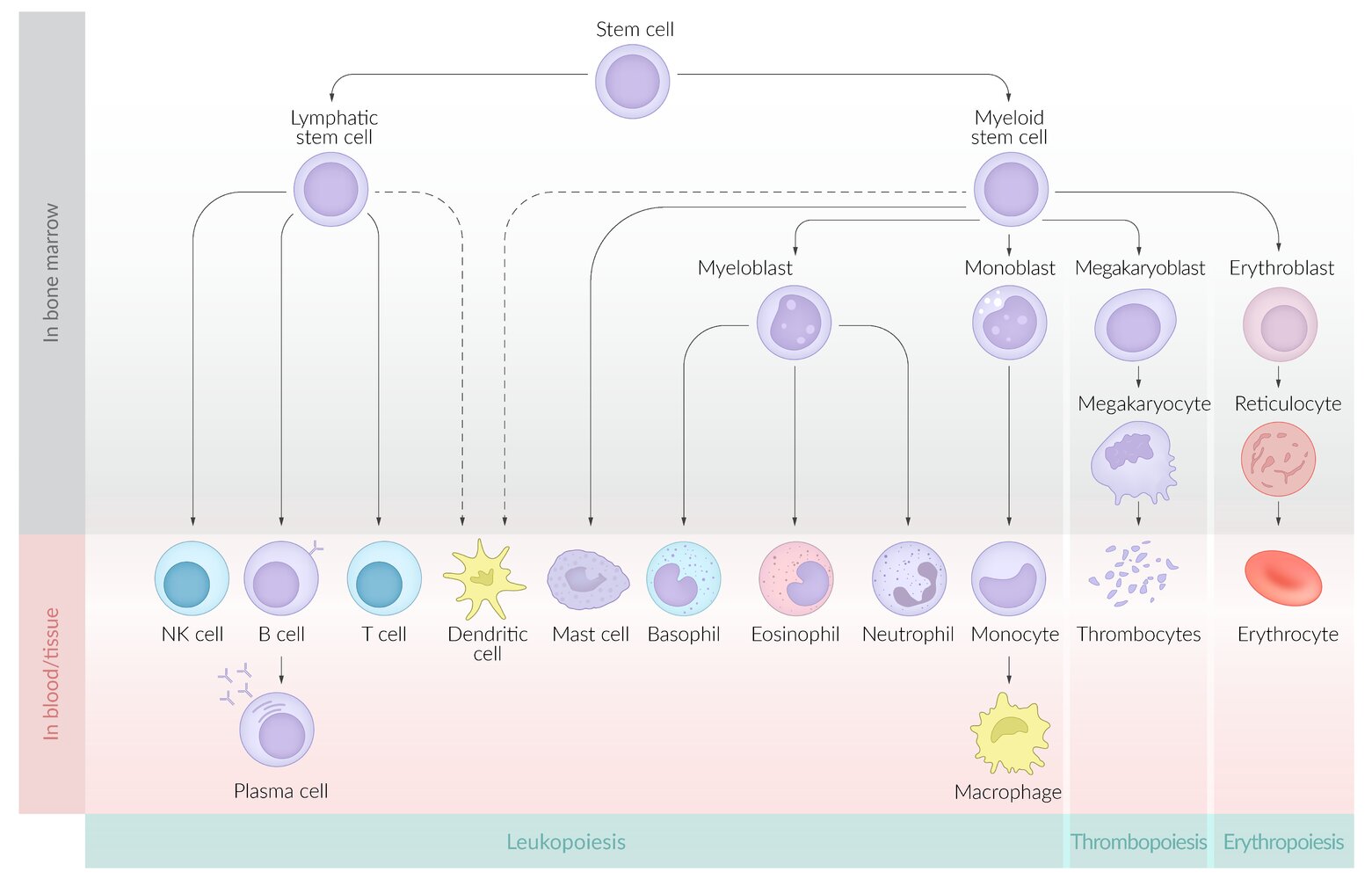
The stages of hematopoiesis, a process by which pluripotent hematopoietic stem cells in the bone marrow differentiate into two main types of precursor cells: myeloid and lymphoid.
Myeloid precursor cells differentiate into erythrocytes, leukocytes, and thrombocytes. Lymphoid precursor cells differentiate into natural killer cells (NK cells) and B and T lymphocytes. Dendritic cells, which are antigen-presenting cells, develop from both myeloid and lymphoid precursors.
© AMBOSS
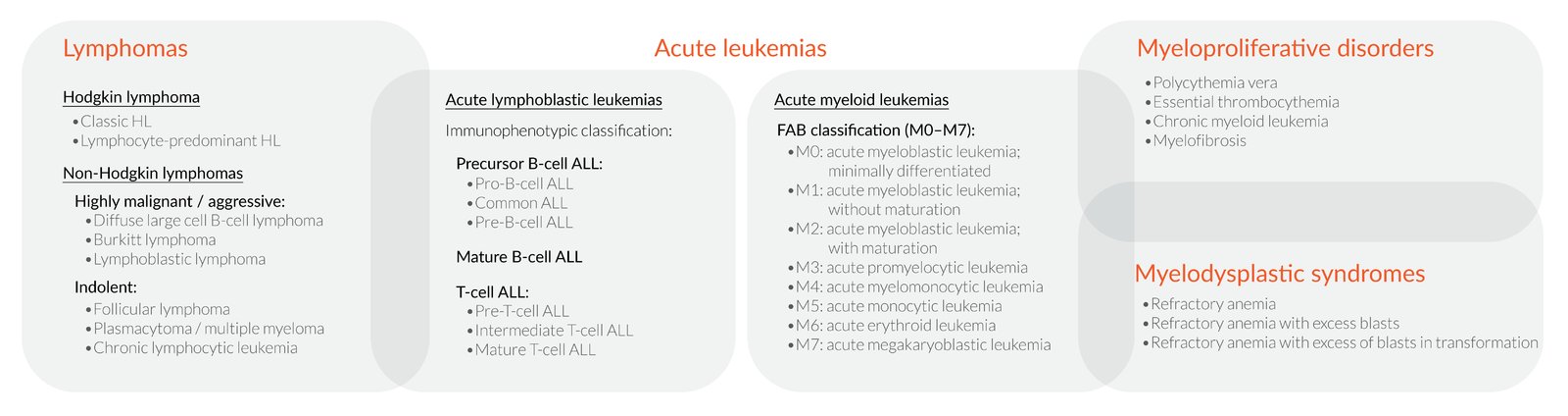
Hematologic neoplasms can be classified according to a variety of overlapping systems, the most important being by:
1. Cell origin
– Lymphatic (lymphomas and lymphoblastic leukemias)
– Myeloid (myeloid leukemias, myeloproliferative disorders, and myelodysplastic syndromes)
2. Disease progression
– Acute (acute leukemias can be either lymphatic or myeloid neoplasms)
– Chronic (e.g., chronic myeloid leukemia)
© AMBOSS
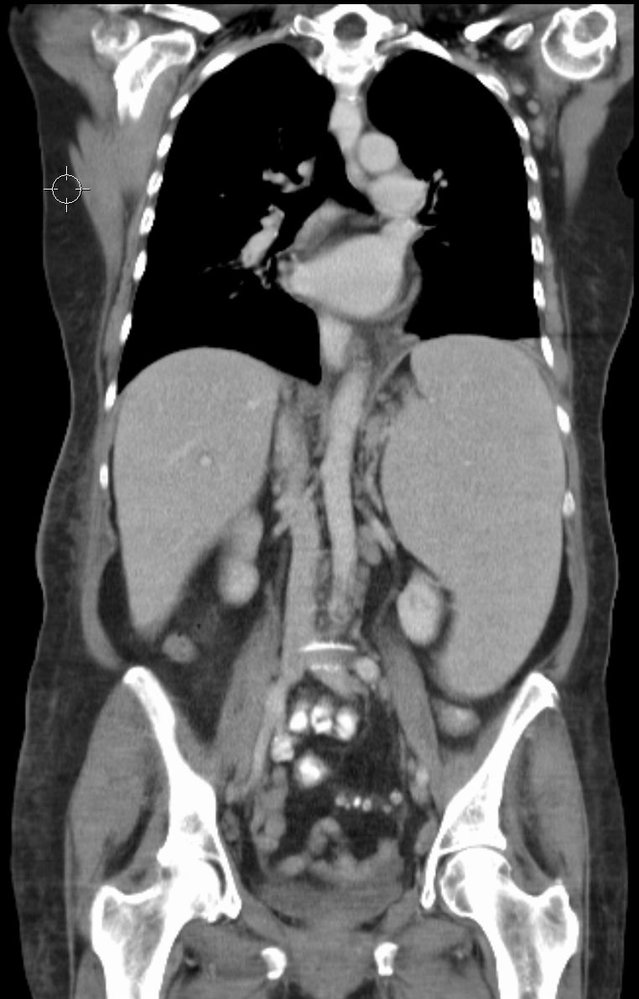
CT scan of the thoracoabdominal cavity (coronal view, with contrast) of a patient with a history of chronic lymphocytic leukemia
The spleen (green overlay) is markedly enlarged. It extends medially (convex toward the midline), inferiorly (into the left iliac fossa), and superiorly (elevating the left hemidiaphragm). The left kidney is displaced inferiorly. The splenic parenchyma shows homogeneous enhancement, without masses or infarcts. Hepatomegaly was also visible on serial CT sections.
A normal spleen measures approx. 12 cm in length. The right hemidiaphragm is typically higher than the left.
A: aorta; IVC: inferior vena cava; K: kidneys; L: liver; PM: psoas muscles
Source: “Splenomegalie bei CLL” by Hellerhoff, Wikimedia Commons, licensed under CC BY-SA 3.0. The supplementary image with overlays of relevant areas was adapted from the image mentioned above and licensed under CC BY-SA 3.0.
{kind=link}
Primary myelofibrosis
Background
-
Description: : any MPN leading to bone marrow fibrosis, extramedullary hematopoiesis, and splenomegaly
- Primary myelofibrosis (also known as agnogenic myeloid metaplasia): occurs spontaneously
- Secondary myelofibrosis: occurs as a result of disease progression in patients previously diagnosed with another MPN, e.g., polycythemia vera (PV), essential thrombocythemia (ET).
- Epidemiology: : Peak incidence is between 50 and 74 years of age. [16]
- Pathophysiology: genetic mutations → hyperplasia of atypical megakaryocytes → ↑ TGF-β → ↑ fibroblast activity → bone marrow obliteration due to fibrosis → displacement of hematopoietic stem cells → extramedullary hematopoiesis
Clinical features [3]
- Constitutional symptoms
- Anemia
- Symptomatic splenomegaly [13]
- Thromboembolic events
- Petechial bleeding
- Increased infections
Diagnostics [1][4]
PMF is a diagnosis of exclusion, while secondary myelofibrosis is typically diagnosed as part of the monitoring of the preceding MPN (e.g., PV, ET). Other causes of bone marrow fibrosis must be ruled out before the diagnosis can be made; see “Differential diagnoses.” [8]
-
Initial studies
-
CBC [3]
- Classic presentation: anemia, thrombocytosis, and leukocytosis
-
Patients may additionally present with:
- Thrombocytopenia (33%)
- Pancytopenia (10%)
- ↑ Leukocyte alkaline phosphatase, LDH, and uric acid
-
CBC [3]
- Peripheral blood smear: : dacrocytes (teardrop cells) and leukoerythroblastosis (overt fibrotic stage)
-
Genetic marker studies [7]
- JAK2 mutation: 50–60% of patients
- CALR mutation: 18–32% of patients
- MPL mutation: 5–9% of patients
-
Bone marrow studies
- Aspiration often fails (dry tap) because of severe marrow fibrosis.
-
Biopsy
- Atypical megakaryocytes
- Prefibrotic stage: hypercellularity (↑ granulopoiesis), possibly ↓ erythropoiesis
- Fibrotic stage: severe fibrosis
As PMF progresses (i.e., overt fibrotic phase), extramedullary hematopoiesis becomes more prominent with signs of pancytopenia and splenomegaly. [1][4]
In myelofibrosis, RBCs shed tears (teardrop cells) because they have been forced out of the fibrosed bone marrow (extramedullary hematopoiesis).

Differential diagnoses [17]
-
Neoplastic conditions
- Other MPNs (e.g., CML, PV, ET)
- Myelodysplastic syndromes
- Lymphomas and leukemias
- Infections (e.g., tuberculosis)
- Endocrine abnormalities (e.g., hyperparathyroidism, vitamin D deficiency)
- Autoimmune diseases (e.g., SLE, Sjogren syndrome, systemic sclerosis, primary autoimmune myelofibrosis)
- Medication side effects (e.g., thrombopoietin receptor agonist toxicity)
Treatment [8][12][18]
Risk scores and screening for high-risk features are used to stratify risk and determine treatment. [19]
- Consider observation for low-risk patients.
- Start treatment in patients with:
- Moderate to high-risk features
- Symptomatic anemia
- Symptomatic splenomegaly
- Secondary myelofibrosis is typically treated with a combination of therapies targeting primary myelofibrosis as well as the preceding MPN (e.g., PV, ET).
High-risk features [19][20]
-
Patient features
- Age ≥ 65 years
- Constitutional symptoms
- Transfusion dependency
-
Laboratory findings
- Anemia (Hgb < 10 g/dL)
- WBC > 25,000/mm3
- Circulating blasts ≥ 1% [13]
- Thrombocytopenia (platelets < 100,000/mm3)
- Unfavorable karyotype
Supportive care
- Thromboembolism prevention: Consider low-dose aspirin [21]
- Symptomatic splenomegaly: Options include ruxolitinib and hydroxyurea (see “Treatment overview” for further information).
- Symptomatic cytopenias: Transfusions are the mainstay of treatment; other options, including thalidomide, may be considered (see “Treatment overview” for further information). [3]
Disease-specific therapy
- Targeted therapy: JAK2 inhibitors (e.g., ruxolitinib) [8][22]
- Allogeneic hematopoietic stem cell transplantation: potentially curative; (typically performed in younger patients with high-risk features and/or a poor prognosis) [8]
Complications [8]
- Progressive bone marrow failure
- Portal hypertension due to splenomegaly
- Pulmonary hypertension
- Transformation to acute leukemia
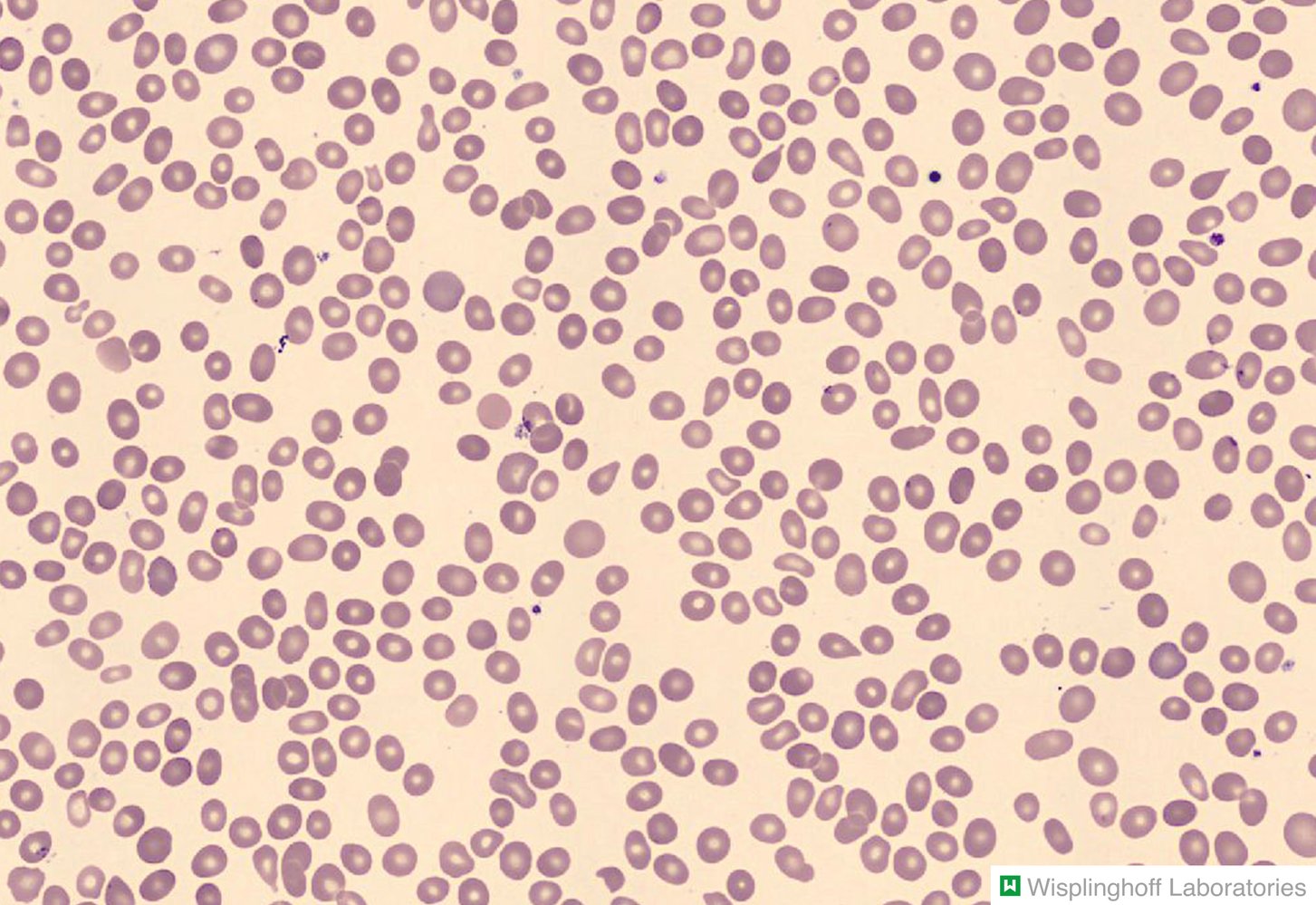
Photomicrograph of a peripheral blood smear
Erythrocytes with a teardrop shape can be seen throughout the image (red overlay). The majority of the erythrocytes appear round and normal.
Teardrop-shaped erythrocytes, known as dacrocytes, are found in conditions that involve extramedullary hematopoiesis (e.g., myelofibrosis, thalassemia, splenomegaly).
Our great thanks to Dr. Wisplinghoff (Dr. Wisplinghoff's laboratory) for kindly providing this image.
Essential thrombocythemia
Background
- Description: isolated uncontrolled proliferation of platelets not caused by another condition (e.g., reactive thrombocytosis, another MPN)
-
Epidemiology [23]
- Median age at diagnosis: 60 years
- ♀ > ♂ (2:1)
- Pathophysiology: genetic mutations → activation of the thrombopoietin receptor → proliferation of platelets [6]
Clinical features [8]
- Commonly asymptomatic
- Thromboembolic events
- Increased risk of fetal loss [3]
- Vasomotor symptoms (headache, visual disturbances, acral paresthesias, ocular migraines)
- Acute gouty arthritis
- Petechial bleeding
- Erythromelalgia


Diagnostics [1][4]
ET is a diagnosis of exclusion and requires ruling out other causes of elevated platelets, such as reactive thrombocytosis and other MPNs (e.g., prefibrotic myelofibrosis, PV). [8]
-
Initial studies
- CBC: isolated sustained thrombocytosis (> 450,000/mm3)
- ↑ LDH and uric acid
- Pseudohyperkalemia [8]
- Peripheral blood smear: large hypogranular platelets
-
Genetic marker studies [7]
- JAK2 mutation: 50–60% of patients
- CALR mutation: 26% of patients
- MPL mutation: 4% of patients
- Bone marrow studies: hyperplasia of mature megakaryocytes
Isolated thrombocytosis is characteristic of ET; however, it may be also seen in early PV and early PMF. [8]
Differential diagnoses
- Reactive thrombocytosis
- Other MPNs with elevated platelets (e.g., prefibrotic PMF, PV, CML)
- Hereditary thrombocytosis
- See also “Thrombocytosis.”
Treatment [8][12][24]
Risk scores and screening for high-risk features are used to stratify risk and determine treatment. [19][25]
Low and intermediate-risk patients
- Observation
- Consider low-dose aspirin. [8]
High-risk patients
Treat in order to prevent thrombohemorrhagic events.
- Start low-dose aspirin [8][26]
- Reduce platelet count with one or more of the following:
- Hydroxyurea
- Anagrelide [27]
- Interferon alpha
- Features that increase the risk of thrombohemorrhagic events include: [19][28]
-
Thrombotic risk factors
- Age ≥ 60 years
- History of thrombosis
- Presence of JAK2 V617F mutation
- Cardiovascular risk factors, e.g., diabetes, hypertension, history of smoking [28]
- Leukocytosis > 11,000/mm3
- Bleeding risk factors
- Platelet count 1,000,000–1,500,000/mm3 is associated with acquired von Willebrand disease (AVWD). [29]
- History of major bleeding
-
Thrombotic risk factors
Complications [8]
- Venous or arterial clots
- AVWD [30][31]
- Transformation to:
- Myelofibrosis
- Acute myeloid leukemia
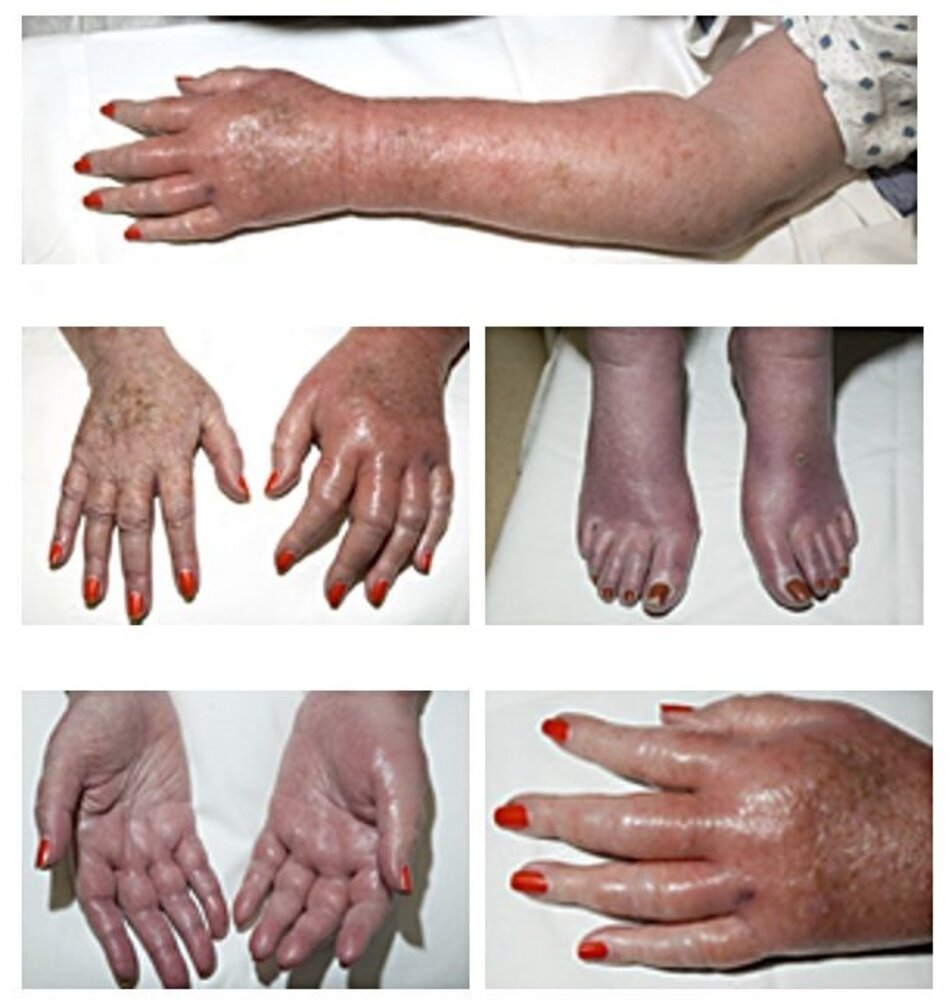
The left upper extremity and both feet appear erythematous and swollen.
The appearance was accompanied by increased skin temperature and burning pain.
Source: “Case 151, in: Images of Memorable Cases: 50 Years at the Bedside” by Fred HL, van Dijk HA, Images of Memorable Cases: 50 Years at the Bedside, licensed under CC BY 2.0.
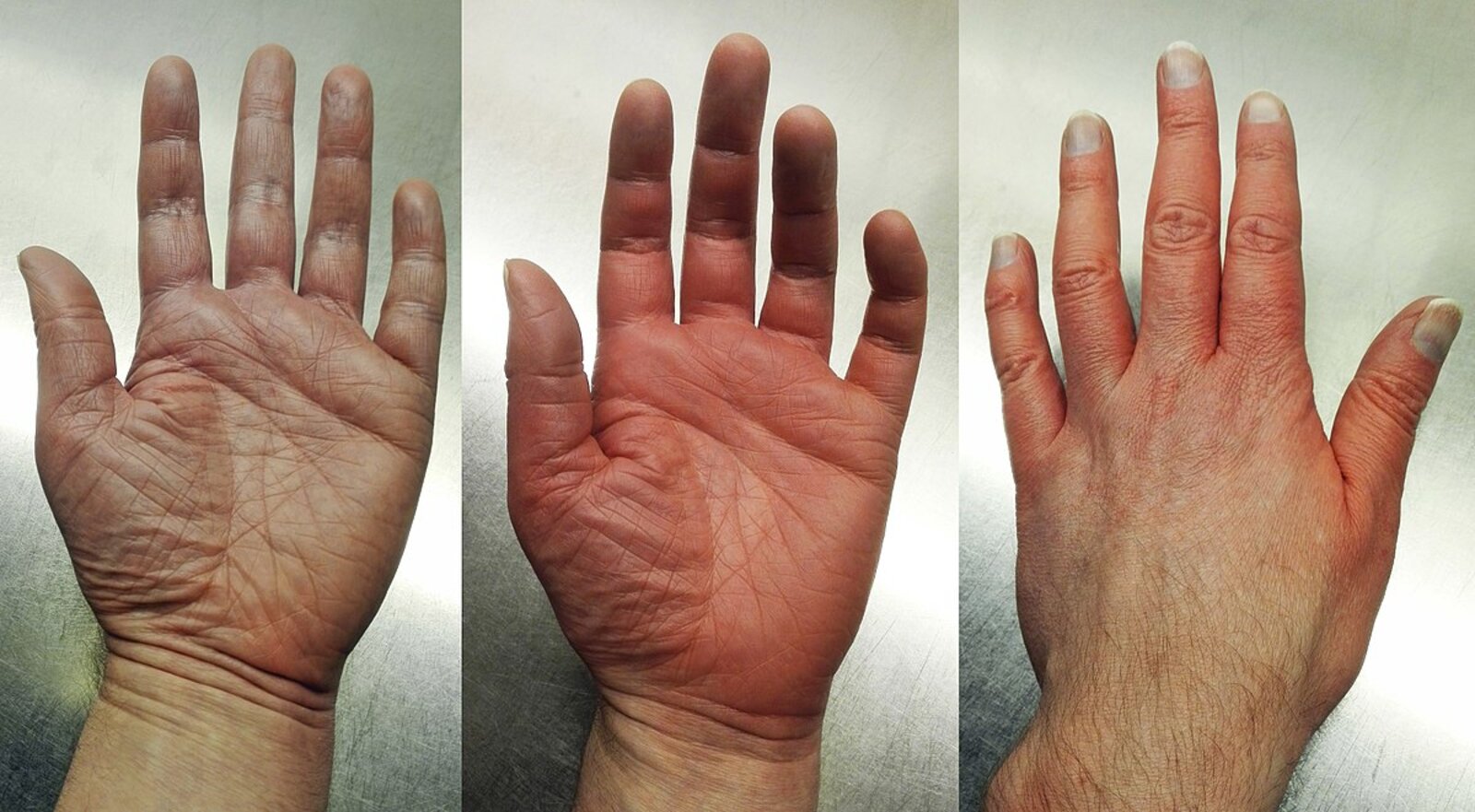
– Left image (just prior to episode of erythromelalgia): Skin is mottled.
– Middle image (during episode of erythromelalgia): Skin is hyperemic, accompanied by increased skin temperature and burning pain.
– Right image (during episode of erythromelalgia): Dorsal aspects of fingers are also erythematous, but fingernails are greyish-white, indicating vasoconstriction (uncommon combination of features, but typical for this particular patient's episodes).
Source: “Erythromelalgia in left hand.jpg” by Minor, Wikimedia Commons, licensed under CC BY-SA 4.0.
{kind=link}
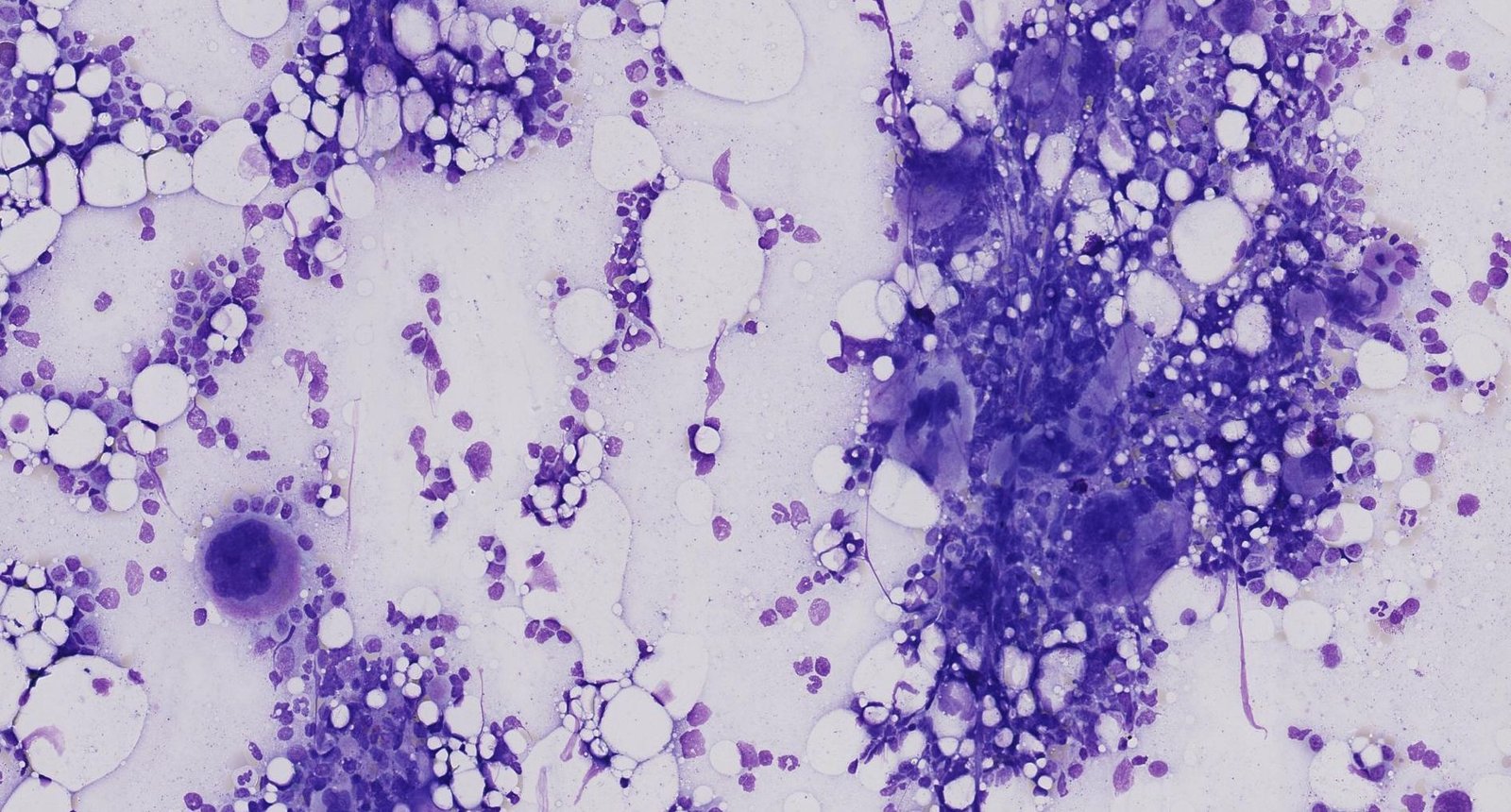
Photomicrograph of a bone marrow aspirate smear (Giemsa stain; high magnification)
The bone marrow appears hypercellular. Giant mature megakaryocytes, identifiable by their large size and multilobulated nucleus (example indicated by arrowhead), can be seen forming multiple clusters (example indicated by yellow overlay).
These are the typical features of essential thrombocythemia (also known as “essential thrombocytosis”), a myeloproliferative neoplasm.
Click on the Smartzoom button to view the entire specimen through a virtual microscope.
Source: © Smart Zoom, Smart In Media. Image and annotations in digital microscopy: Prof. Dr. Karl-Anton Kreuzer, Facharzt für Innere Medizin, Hämatologie und Onkologie
Chronic eosinophilic leukemia
- Description: leukemia with monoclonal proliferation of eosinophilic granulocytes that causes peripheral eosinophilia and tissue damage
- Etiology: various associated cytogenetic mutations, particularly translocations of chromosome 5 and trisomy 8 [3]
-
Clinical features [3]
- Hepatosplenomegaly
- Anemia; and/or coagulation disorders
- Cardiac, pulmonary, hepatic, splenic, and CNS involvement
-
Diagnosis
- CBC: ↑↑↑ eosinophils (> 1,500/mm3) [4]
- Peripheral blood smear and bone marrow aspiration: monoclonal blast cells or cytogenetically abnormal eosinophils
-
Treatment [3]
- Tyrosine kinase inhibitors, e.g., imatinib
- Allogeneic stem cell transplantation
- Symptomatic relief to decrease eosinophil counts, e.g., glucocorticoids, hydroxyurea, or antiinterleukin-5 therapy

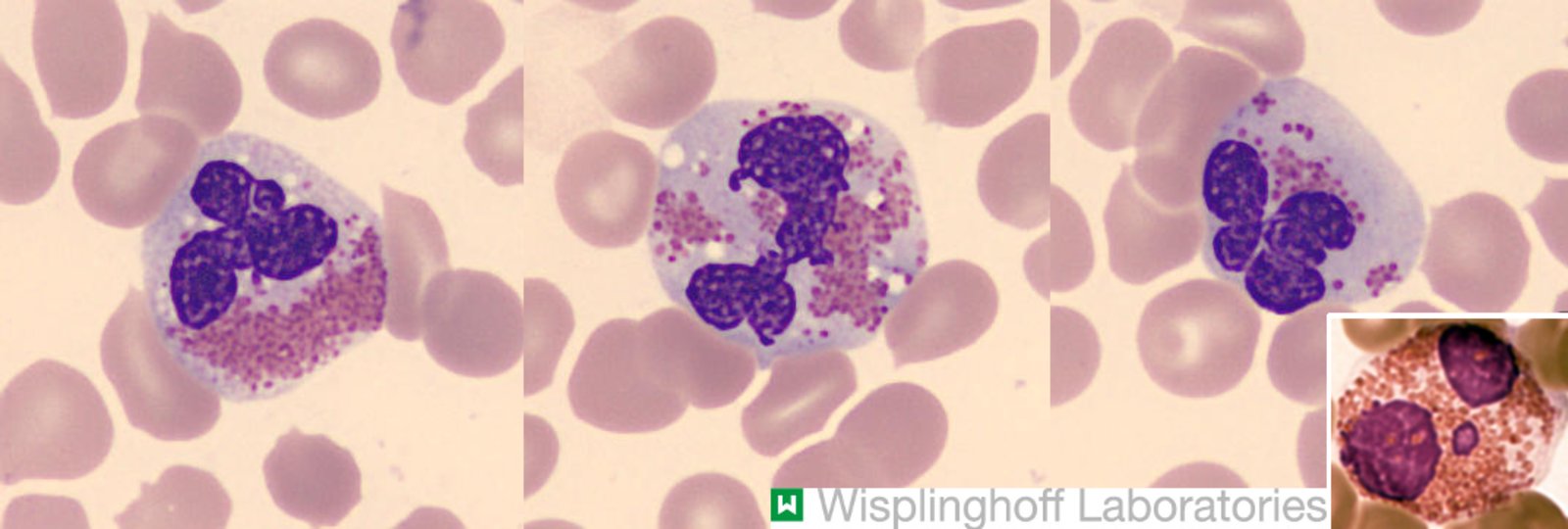
Photomicrographs of peripheral blood smear (Pappenheim stain; very high magnification)
The three large images show eosinophilic granulocytes with pathological changes, including nuclear atypia (hyperlobation; white arrows), degranulation (granule-free cytoplasmic areas; green overlay), and vacuole formation (dark gray overlay).
In combination with eosinophilia, these findings are typical of chronic eosinophilic leukemia, although they are also seen in reactive conditions.
The small inset shows a normal eosinophilic granulocyte from a different patient for comparison. Note the uniform distribution of granules and the absence of vacuoles or nuclear atypia.
Our great thanks to Dr. Wisplinghoff (Dr. Wisplinghoff's laboratory) for kindly providing this image. Further notes: Modifications: Collage with usage of public domain image. License link: https://en.wikipedia.org/wiki/Public_domain. Source title: Eosinophil and polymorphonuclear neutrophil.jpg; Source link: https://commons.wikimedia.org/wiki/File:Eosinophil_and_polymorphonuclear_neutrophil.jpg. Author of source: Davidcsaba. Source author link: https://commons.wikimedia.org/w/index.php?title=User:Davidcsaba&action=edit&redlink=1. Source designation: Wikimedia Commons. Cropping of named public domain image. Overlay for Collage.
Chronic neutrophilic leukemia
- Definition: an extremely rare MPN characterized by leukocytosis with significant left shift [32][33]
- Etiology: CSF3R gene mutation (most common) [34]
-
Clinical features [3]
- Fatigue
- Anorexia
- Abdominal pain secondary to hepatosplenomegaly
- Symptoms of gouty arthritis
-
Diagnostics [3][4][35]
- CBC: leukocytosis (WBC ≥ 25,000/mm3) with neutrophilia , mild to moderate anemia, and thrombocytopenia
- ↑ LDH, ↑ leukocyte alkaline phosphatase
- Bone marrow: granulocytic hyperplasia with high myeloid:erythroid ratio
-
Treatment [3][35]
- There is no optimal treatment regimen because of the rarity of the condition.
- Patients are often refractory to treatments used for other MPNs.
- Depending on the mutation, targeted therapies may be appropriate (e.g., tyrosine kinase inhibitors).
- Allogeneic stem cell transplantation may be curative.
Myeloproliferative neoplasm, unclassifiable
- Definition: an MPN that does not meet the WHO diagnostic criteria for any other MPN [36]
- Etiology: unclear; cytogenic mutations may occur but do not involve classic mutations associated with other MPNs. [3]
-
Clinical features [36][37]
- Constitutional symptoms
- Pruritus
- Hepatosplenomegaly (advanced disease)
- Unexplained thrombosis, particularly splanchnic vein thrombosis
-
Diagnostics [3][37]
- CBC: mild to marked leukocytosis, which may be accompanied by anemia and/or thrombocytosis
- ↑ LDH
- Bone marrow: hypercellularity, clustered pleomorphic megakaryocytes [36]
-
Treatment [3]
- Typically symptomatic relief and prevention of thrombosis (see “Treatment overview”)
- Allogeneic stem cell transplantation may be curative.
External Resources
References
- Arber DA, Orazi A, Hasserjian R, et al. "The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia". Blood. 127(20). :2391-2405. (2016)
- Mannelli F. "Acute Myeloid Leukemia Evolving from Myeloproliferative Neoplasms: Many Sides of a Challenging Disease". J Clin Med. 10(3). :436. (2021)
- Caligiuri M, Levi MM, Kaushansky K, et al. "Williams Hematology, 9E". McGraw-Hill Education / Medical. (2015). ISBN: 9780071833004
- Barbui T, Thiele J, Gisslinger H, et al. "The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion". Blood Cancer J. 8(2). :15. (2018)
- Er T-K, Lin S-F, Chang J-G, et al. "Detection of the JAK2 V617F missense mutation by high resolution melting analysis and its validation". Clin Chim Acta. 408(1-2). :39-44. (2009)
- Shammo JM, Stein BL. "Mutations in MPNs: prognostic implications, window to biology, and impact on treatment decisions". Hematology Am Soc Hematol Educ Program. 2016(1). :552-560. (2016)
- Szybinski J, Meyer SC. "Genetics of Myeloproliferative Neoplasms". Hematol Oncol Clin North Am. 35(2). :217-236. (2021)
- Spivak JL. "Myeloproliferative Neoplasms". N Engl J Med. 376(22). :2168-2181. (2017)
- Murphy S. "Polycythemia vera". Dis Mon. 38(3). :158-212. (1992)
- Shibusawa M, Tadokoro J, Kojima M, Kashimura M. "Chronic myelogenous leukaemia with a p53 mutation demonstrated neutrophilic granulocytes with nuclear hypolobation (pseudo-Pelger-Hüet anomaly) and hypogranulation in the peripheral blood smear". BMJ Case Rep. :bcr-2017-221907. (2018)
- Patel AA, Odenike O. "Genomics of MPN progression". Hematology. 2020(1). :440-449. (2020)
- Vannucchi AM, Harrison CN. "Emerging treatments for classical myeloproliferative neoplasms". Blood. 129(6). :693-703. (2017)
- Mesa RA. "How I treat symptomatic splenomegaly in patients with myelofibrosis". Blood. 113(22). :5394-5400. (2009)
- Luo X, Xu Z, Li B, et al. "Thalidomide plus prednisone with or without danazol therapy in myelofibrosis: a retrospective analysis of incidence and durability of anemia response". Blood Cancer J. 8(1). (2018)
- Al-Sharefi A, Mohammed A, Abdalaziz A, Jayasena CN. "Androgens and Anemia: Current Trends and Future Prospects". Front Endocrinol (Lausanne). 10. (2019)
- Accurso V, Santoro M, Mancuso S, et al. "The Essential Thrombocythemia in 2020: What We Know and Where We Still Have to Dig Deep". Clinical Medicine Insights: Blood Disorders. 13. :263485352097821. (2020)
- Birgegård G, Besses C, Griesshammer M, et al. "Treatment of essential thrombocythemia in Europe: a prospective long-term observational study of 3649 high-risk patients in the Evaluation of Anagrelide Efficacy and Long-term Safety study". Haematologica. 103(1). :51-60. (2017)
- Rumi E, Cazzola M. "Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms". Blood. 129(6). :680-692. (2017)
- Barbui T, Barosi G, Birgegard G, et al. "Philadelphia-Negative Classical Myeloproliferative Neoplasms: Critical Concepts and Management Recommendations From European LeukemiaNet". J Clin Oncol. 29(6). :761-770. (2011)
- Tefferi A. "Myeloproliferative neoplasms: A decade of discoveries and treatment advances". Am J Hematol. 91(1). :50-58. (2015)
- Birgegård G. "The Use of Anagrelide in Myeloproliferative Neoplasms, with Focus on Essential Thrombocythemia". Curr Hematol Malig Rep. 11(5). :348-355. (2016)
- Haider M, Gangat N, Lasho T, et al. "Validation of the revised international prognostic score of thrombosis for essential thrombocythemia (IPSET-thrombosis) in 585 Mayo clinic patients". Am J Hematol. 91(4). :390-394. (2016)
- Tefferi A, Vannucchi AM, Barbui T. "Essential thrombocythemia treatment algorithm 2018". Blood Cancer Journal. 8(1). (2018)
- Michiels JJ. "Acquired von Willebrand Disease Due to Increasing Platelet Count Can Readily Explain the Paradox of Thrombosis and Bleeding in Thrombocythemia". Clinical and Applied Thrombosis/Hemostasis. 5(3). :147-151. (1999)
- Awada H, Voso M, Guglielmelli P, Gurnari C. "Essential Thrombocythemia and Acquired von Willebrand Syndrome: The Shadowlands between Thrombosis and Bleeding". Cancers. 12(7). :1746. (2020)
- Benjamin Garmezy, Jordan K. Schaefer, Jessica Mercer, Moshe Talpaz. "A provider's guide to primary myelofibrosis: pathophysiology, diagnosis, and management". Blood Rev. 45. :100691. (2021)
- Marcellino B, El Jamal SM, Mascarenhas JO. "Distinguishing autoimmune myelofibrosis from primary myelofibrosis.". Clin Adv Hematol Oncol. 16(9). :619-626. (2018)
- De Melo Campos P. "Primary myelofibrosis: current therapeutic options". Rev Bras Hematol Hemoter. 38(3). :257-263. (2016)
- Gangat N, Caramazza D, Vaidya R, et al. "DIPSS Plus: A Refined Dynamic International Prognostic Scoring System for Primary Myelofibrosis That Incorporates Prognostic Information From Karyotype, Platelet Count, and Transfusion Status". J Clin Oncol. 29(4). :392-397. (2011)
- Finazzi G, Vannucchi AM, Barbui T. "Prefibrotic myelofibrosis: treatment algorithm 2018". Blood Cancer J. 8(11). (2018)
- "JAKAFI® (ruxolitinib)". https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/202192Orig1s019Rpllbl.pdf. [2020-01-01]
- Bain BJ, Ahmad S. "Chronic neutrophilic leukaemia and plasma cell-related neutrophilic leukaemoid reactions". Br J Haematol. 171(3). :400-410. (2015)
- Gotlib J, Maxson JE, George TI, Tyner JW. "The new genetics of chronic neutrophilic leukemia and atypical CML: implications for diagnosis and treatment". Blood. 122(10). :1707-1711. (2013)
- Szuber N, Elliott M, Tefferi A. "Chronic neutrophilic leukemia: 2020 update on diagnosis, molecular genetics, prognosis, and management". Am J Hematol. 95(2). :212-224. (2019)
- Szuber N, Tefferi A. "Chronic neutrophilic leukemia: new science and new diagnostic criteria". Blood Cancer Journal. 8(2). (2018)
- Deschamps P, Moonim M, Radia D, et al. "Clinicopathological characterisation of myeloproliferative neoplasm‐unclassifiable (MPN‐U): a retrospective analysis from a large UK tertiary referral centre". Br J Haematol. 193(4). :792-797. (2021)
- Gianelli U, Cattaneo D, Bossi A, et al. "The myeloproliferative neoplasms, unclassifiable: clinical and pathological considerations". Mod Pathol. 30(2). :169-179. (2016)