Summary
This article provides an overview of inherited symptom complexes that occur rarely in the general population. These syndromes are caused by inherited genetic defects, which occur either due to chromosomal aberrations or autosomal/sex-linked traits. The presentation differs for each syndrome, with most features arising from developmental, functional, or structural anomalies of various organs. The diagnosis can be confirmed with the help of molecular genetic detection, fluorescence in situ hybridization (FISH), or other genetic/chromosomal studies. Treatment is usually symptomatic.
Prader-Willi syndrome and Angelman syndrome
Overview
- Definition: genetic syndromes caused by microdeletion (at 15q11-q13); in combination with genomic imprinting.
-
Etiology: The resulting condition depends on the affected gene copy.
-
Angelman syndrome
- Deletion or mutation of maternal UBE3A (chromosome 15) gene copy and paternal gene methylation (silencing)
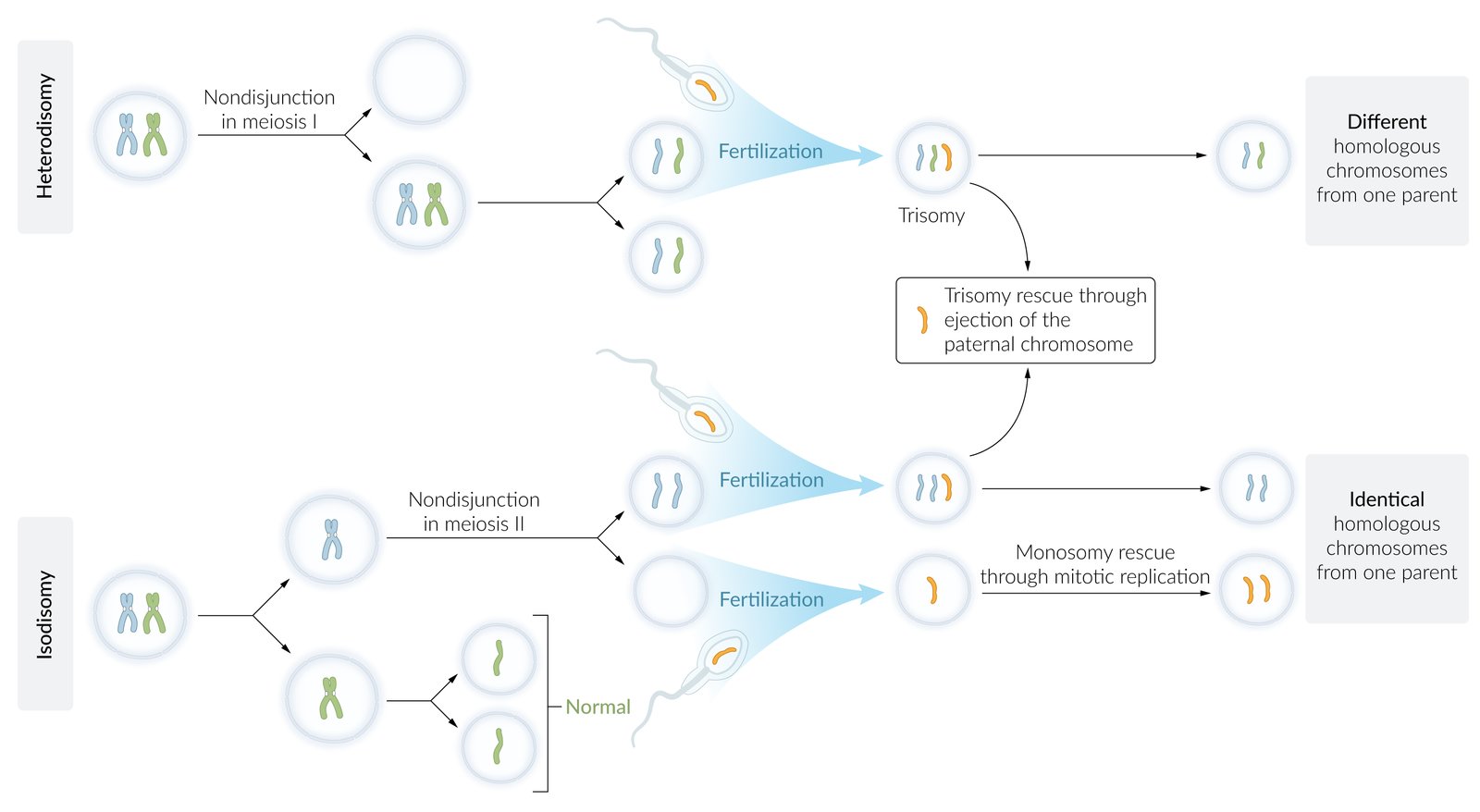
- In ∼ 5% of cases, it results from paternal uniparental disomy (i.e. both copies of chromosome 15 are inherited from the father).
-
Prader-Willi syndrome
- Deletion or mutation of paternal gene copy and maternal gene methylation (silencing)
- Caused by maternal uniparental disomy in about 20–35% of cases [1][2]
-
Angelman syndrome
-
Diagnosis: genetic tests
- Fluorescence in situ hybridization (FISH)
- Methylation testing
“Prader misses his Papa, and Angel her Mama”: allele mutation/deletion of paternal origin in Prader-Willi syndrome and maternal in Angelman syndrome.

Prader-Willi syndrome [3][4][5]
-
Clinical features
- Muscular hypotonia and poor feeding in infants
- Increased appetite (hyperphagia) and obesity
- Short stature , scoliosis
- Cryptorchidism, hypogonadism , genital hypoplasia
- Facial dysmorphia (e.g., almond-shaped eyes, thin upper lip)
- Premature adrenarche with early development of pubic/axillary hair
- Developmental delays; (e.g., delayed achievement of milestones), intellectual disability
- Behavioral problems (e.g., temper tantrums, stubbornness, obsessive-compulsive behavior)
-
Treatment
- Calorie restriction
- Substitution of growth hormone and sex hormones
-
Complications
- Sleep apnea (most common)
- Type 2 diabetes mellitus
- Choking episodes
- Gastric distention and rupture
- Prognosis: normal life expectancy if extreme obesity is avoided


Angelman syndrome [5][6][7]
-
Clinical features
- Delayed mental development and acquisition of motor skills in infants and young children; absent speech development
- Intellectual disability
- In more than 80% of cases, pronounced epileptic seizures
- Microcephaly
- Ataxia, tremulous movements of the limbs
- Truncal hypotonia, limb hypertonia, hyperreflexia
- Difficulty sleeping
- Characteristic happy demeanor with frequent laughing (inappropriate laughter)
- Hyperexcitability, short attention span
- Fascination with water
-
Treatment
- No specific treatment
- Physical, occupational, and speech therapy
- Antiepileptic drugs (if applicable)
- Prognosis: Life expectancy is typically normal.


“Angels in the HEAVENS”: Happy-go-lucky, Easily Excitable personality, Ataxia, Verbal underdevelopment, Epileptic seizures, abNormal facial features, Severe intellectual disability.
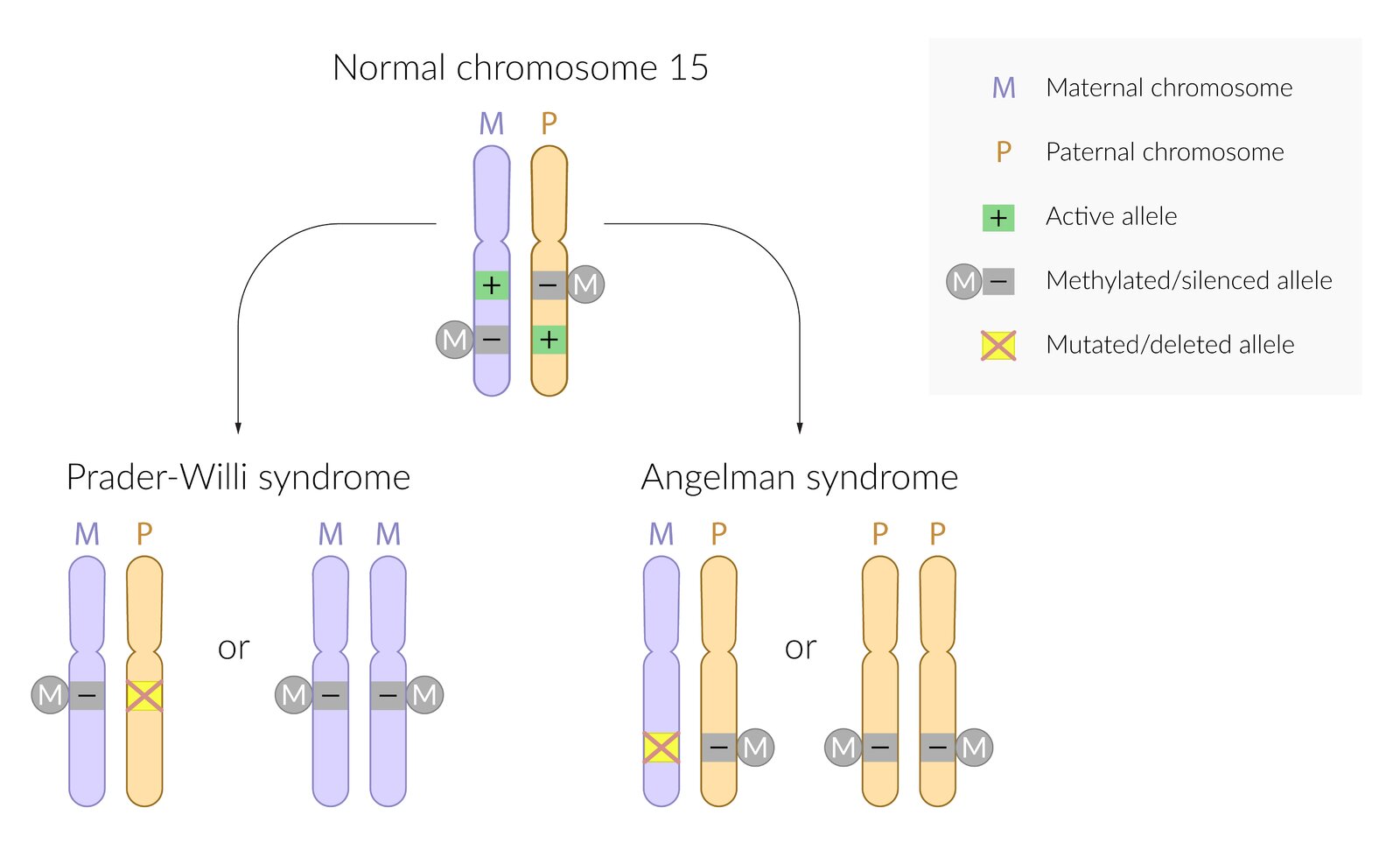
© AMBOSS
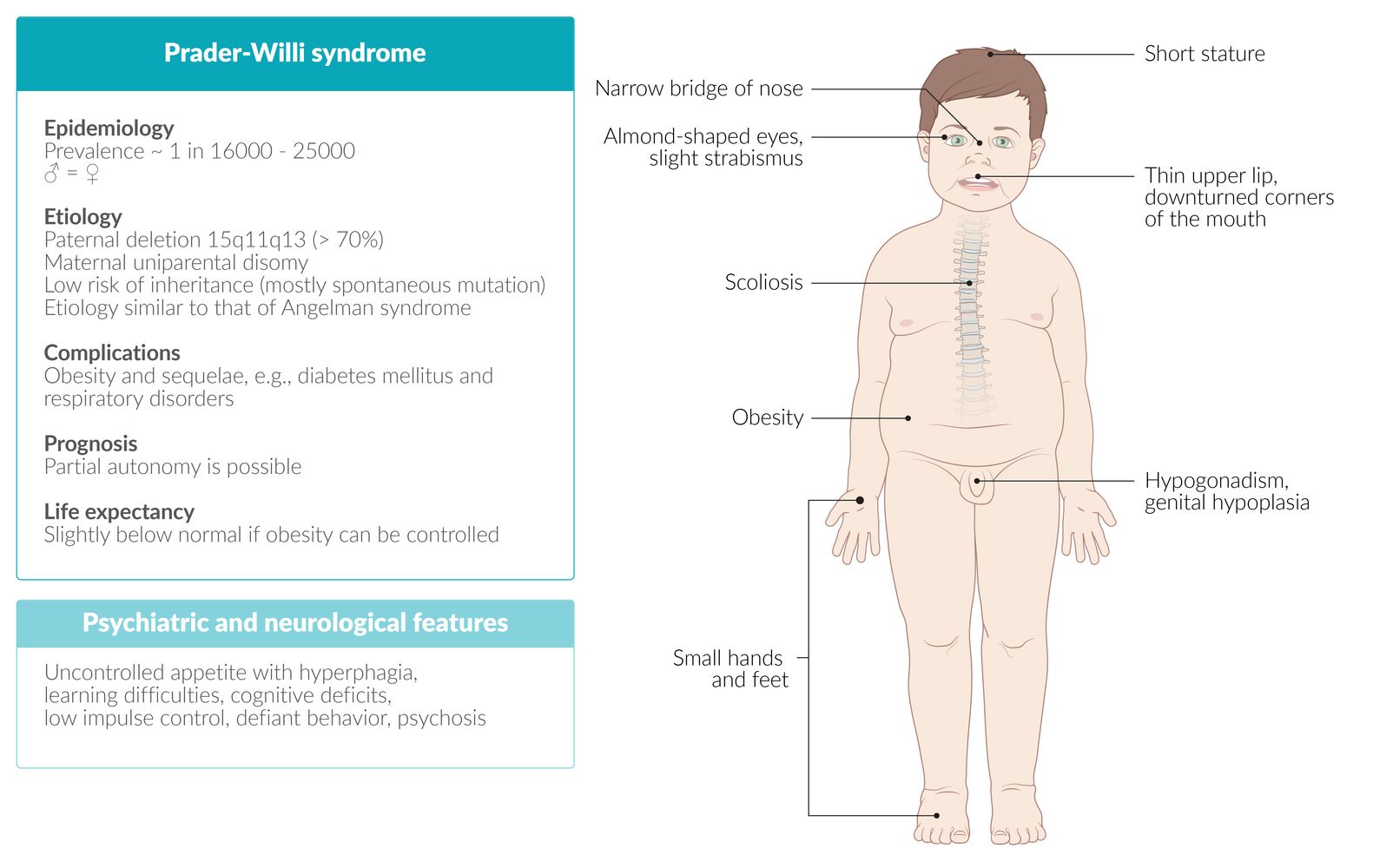
© AMBOSS
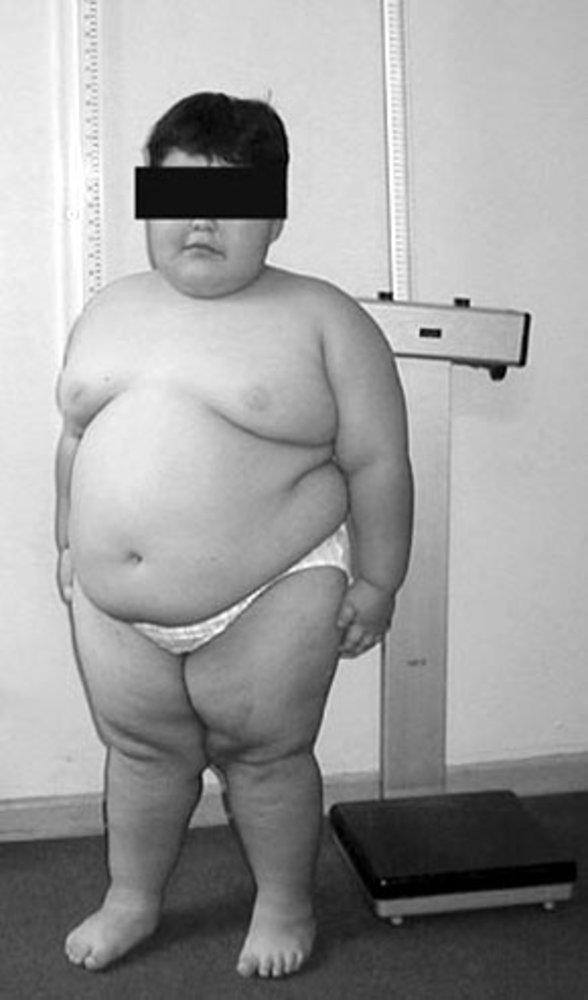
Child of primary school age with massive obesity and a short stature. Both features are typical in children with Prader-Willi syndrome.
Source: “Figure 1. in: Caracterización clínico-genético-molecular de 45 pacientes chilenos con Síndrome de Prader Willi” by Fanny Cortés M et al., Revista Medica De Chile Journal, licensed under CC BY 4.0.
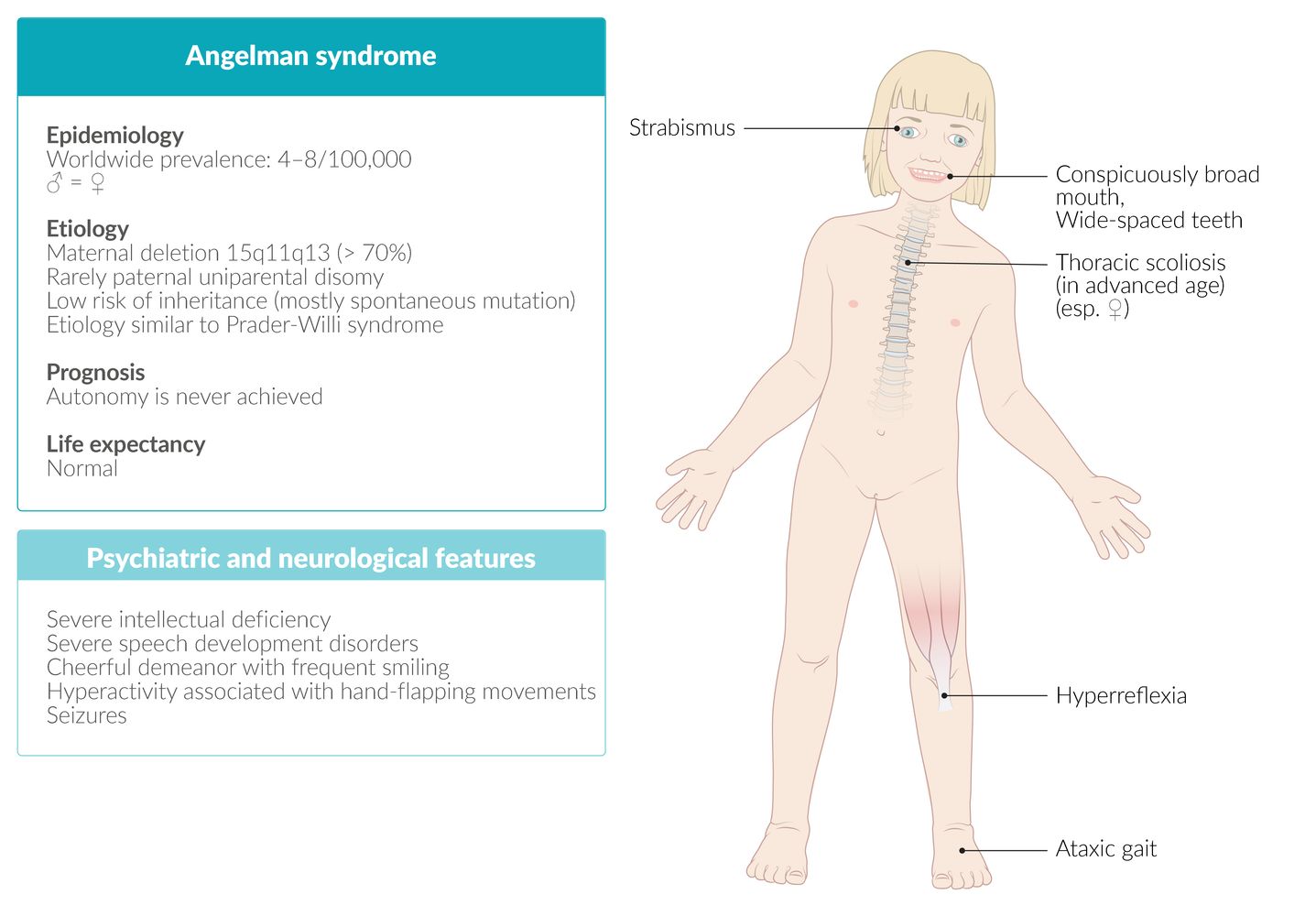
© AMBOSS
© AMBOSS
Fragile X syndrome
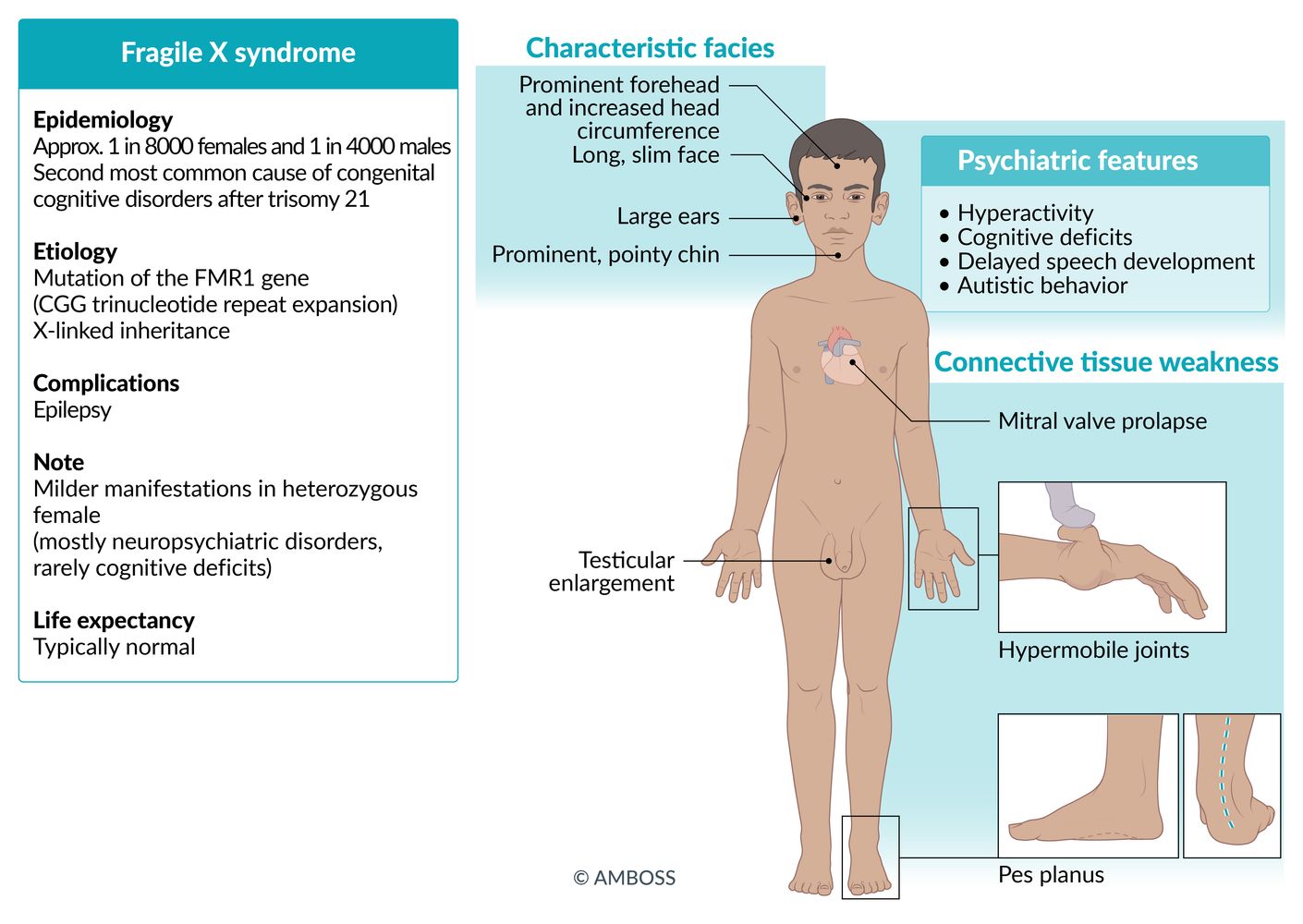
- Definition: an X-linked dominant disease caused by a CGG trinucleotide repeat expansion in the FMR1 gene (fragile X mental retardation 1 gene) during oogenesis. This leads to its hypermethylation, which silences the gene and renders it unable to express its physiological gene product. [8][9]
-
Epidemiology
- Second most common genetic cause of intellectual disability (after trisomy 21)
- Most common inherited cause of intellectual disability, as trisomy 21 mostly occurs sporadically
- Sex distribution: ♂ >> ♀
-
Clinical features: The clinical presentation varies depending on the number of trinucleotide repeats.
- 50–200 repeats (premutation): ataxia, primary ovarian insufficiency, tremor
-
> 200 repeats (full mutation):
- Intellectual disability of varying severity
- Delayed language development
- Behavioral features: autistic behavior, hyperactivity, anxiety
-
Characteristic facial anomalies
- Long and narrow face
- Prominent forehead and jaw
- Large everted ears
- Hypermobile joints
- In men: postpubertal macroorchidism (enlarged testes; rarely occurs prior to puberty)
- Mitral valve prolapse: can lead to mitral regurgitation
- Above-average head circumference
- Focal seizures
-
Diagnosis
- Molecular genetic detection (PCR, Southern blot)
- Cytogenetic detection (not very sensitive)
- Echocardiography
- Treatment: symptomatic
- Prognosis: generally normal life expectancy
Fragile X: “X-tra large” ears, testes, and face in these patients.
© AMBOSS
Macrocephaly and large earlobes.
Source: “File:Fragx-2.jpg” by Peter Saxon, Wikimedia Commons, licensed under CC BY-SA 4.0. Modifications: Eyes pixelated.
{kind=link}

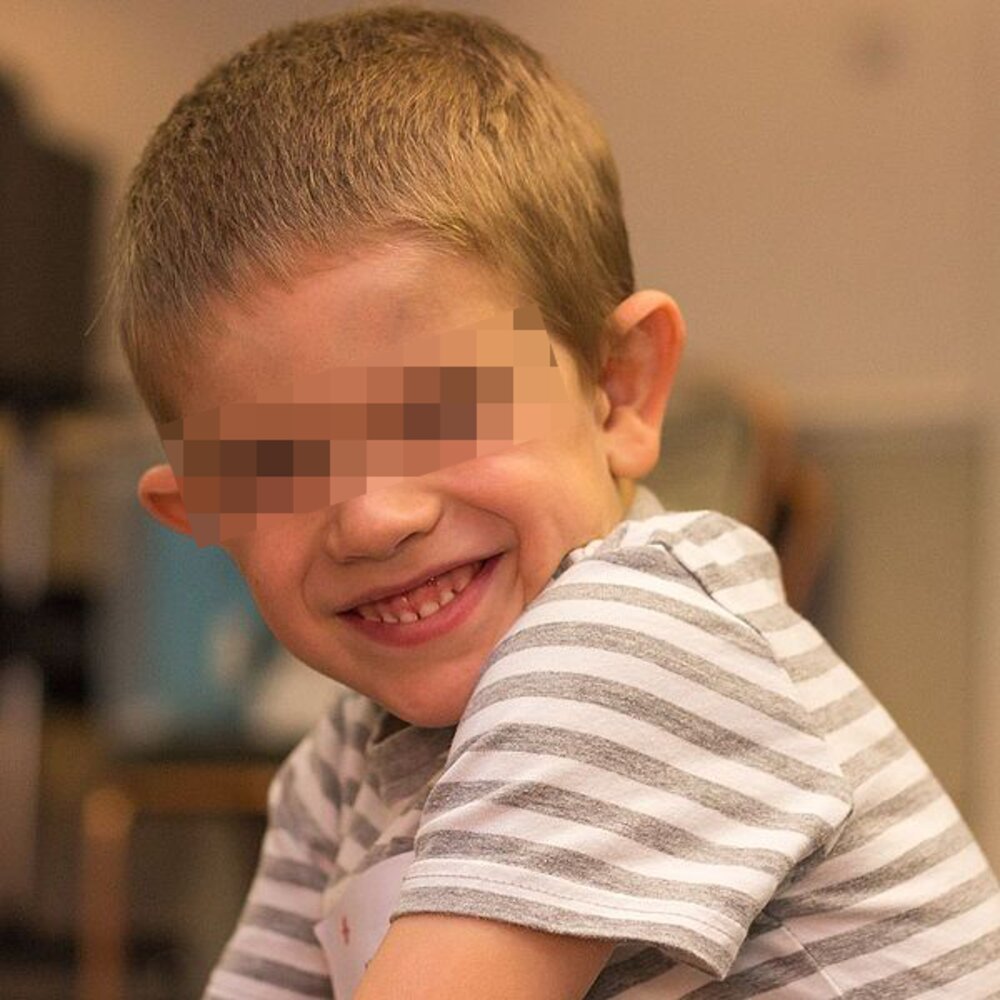
Boy of primary school age with a long and narrow face and large ears. Both features are typical in children with fragile X syndrome.
Source: “Fragx-1” by Peter Saxon, Wikimedia Commons, licensed under CC BY-SA 4.0. Modifications: eyes blurred.
{kind=link}

Original title: “Genetics: Section 9.3 - Fragile X Syndrome”. Created by: Physeo™.
Rett syndrome
- Definition: X-linked disorder with progressive loss of intelligence and cognitive abilities such as language, locomotion, and fine motor skills [10]
-
Etiology: X-linked dominant gene mutation in methyl-CpG binding protein 2 gene (MECP2 gene)
- Usually not an inherited gene defect, but rather a sporadic mutation
- Mutation usually occurs in the paternal allele; thus, females are almost exclusively affected
- If affected, male fetuses die in utero or shortly after birth.
-
Clinical features
- Normal development until the first symptoms appear, which typically happens at 6–18 months of age
-
Symptoms of neurodevelopmental regression, including:
- Loss of motor skills, especially targeted hand movements (affected children show characteristic hand wringing)
- Truncal ataxia, apraxia, choreatic movements
- Intellectual and verbal disability
- Seizures
- Growth failure
- Scoliosis
- Hyperventilation
- Microcephaly
- Diagnosis: : a combination of typical clinical presentation and gene mutation detection
- Prognosis: There is not enough data regarding life expectancy beyond the age of 40, as long-term studies are not available and the disease is fairly rare.
Rett Regresses: Normal development is shortly followed by a loss of targeted hand movements and intellectual and verbal disability.


Original title: “Genetics: Section 7.2 - Rett Syndrome”. Created by: Physeo™.
Cri-du-chat syndrome (cat cry syndrome)
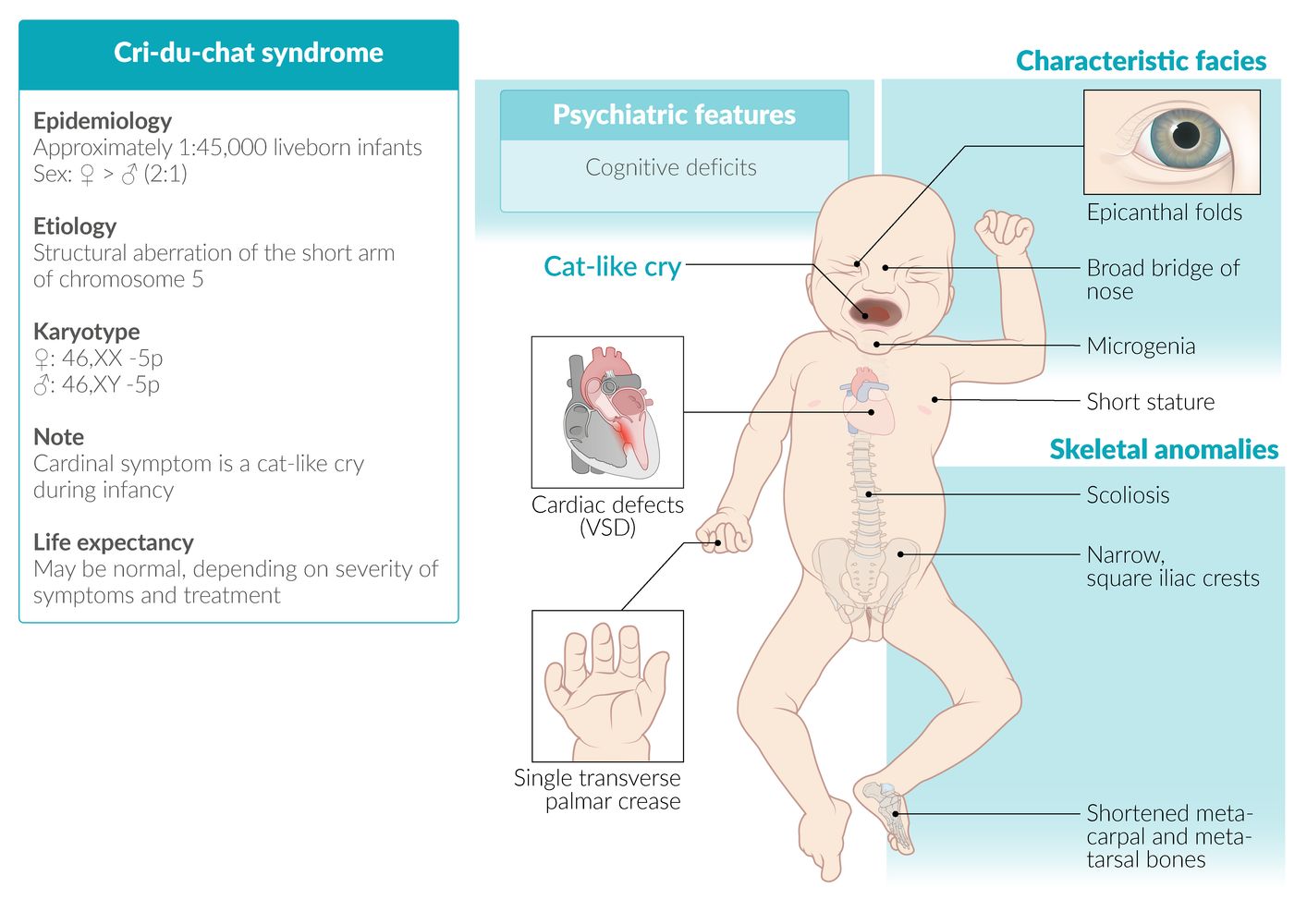
- Definition: rare syndrome caused by a chromosome 5 aberration [11][12]
- Epidemiology: sex distribution: ♀ > ♂ (2:1)
- Etiology: microdeletion of the short arm at chromosome 5 (46,XX,5p- or 46,XY, 5p‑)
-
Clinical features
- Cat-like, high-pitched crying in affected infants
- Congenital heart defects, e.g., VSD
- Microcephaly
- Intellectual disability (moderate to severe)
- Single palmar crease
-
Dysmorphic facial features
- Moon facies
- Widely spaced eyes
- Epicanthal folds
- Broad nasal bridge
- Downward-slanting palpebral fissures
- Skeletal abnormalities
-
Treatment
- Symptomatic treatment
- Early psychological and physical assistance
- Prognosis: A normal life expectancy is possible, but depends on the accompanying symptoms and therapeutic assistance.
 fact sheet")
© AMBOSS

Original title: “Genetics: Section 12.1 - Cri-Du-Chat Syndrome”. Created by: Physeo™.
Williams syndrome
- Definition: a multisystem developmental disorder caused by a deletion at chromosome 7
- Etiology: : microdeletion of the long arm of chromosome 7 (includes a deletion of the elastin gene)
-
Clinical features
- Intellectual disability
- Hypersociability (e.g., comfort with strangers), good verbal skills
-
Characteristic facial features (elfin facies):
- Midfacial hypoplasia,
- Short palpebral fissure
- Wide forehead
- Flattened nasal bridge, anteverted nostrils
- Long philtrum
- Hypodontia
- Cardiovascular malformations; (esp. supravalvular aortic stenosis, renal artery stenosis)
- Hypercalcemia (due to increased sensitivity to vitamin D)
- Low muscle tone
- Hyperacusis, phonophobia
- Potential malformations and development problems in multiple organ systems, such as starburst-like pattern in the iris ("stellate iris")
- Diagnosis: : a combination of typical clinical presentation and gene mutation detection


William takes ICEcream from strangers (Intellectual disabilities, Cardiovascular malformations, Elfin-like facial features, comfort with strangers).
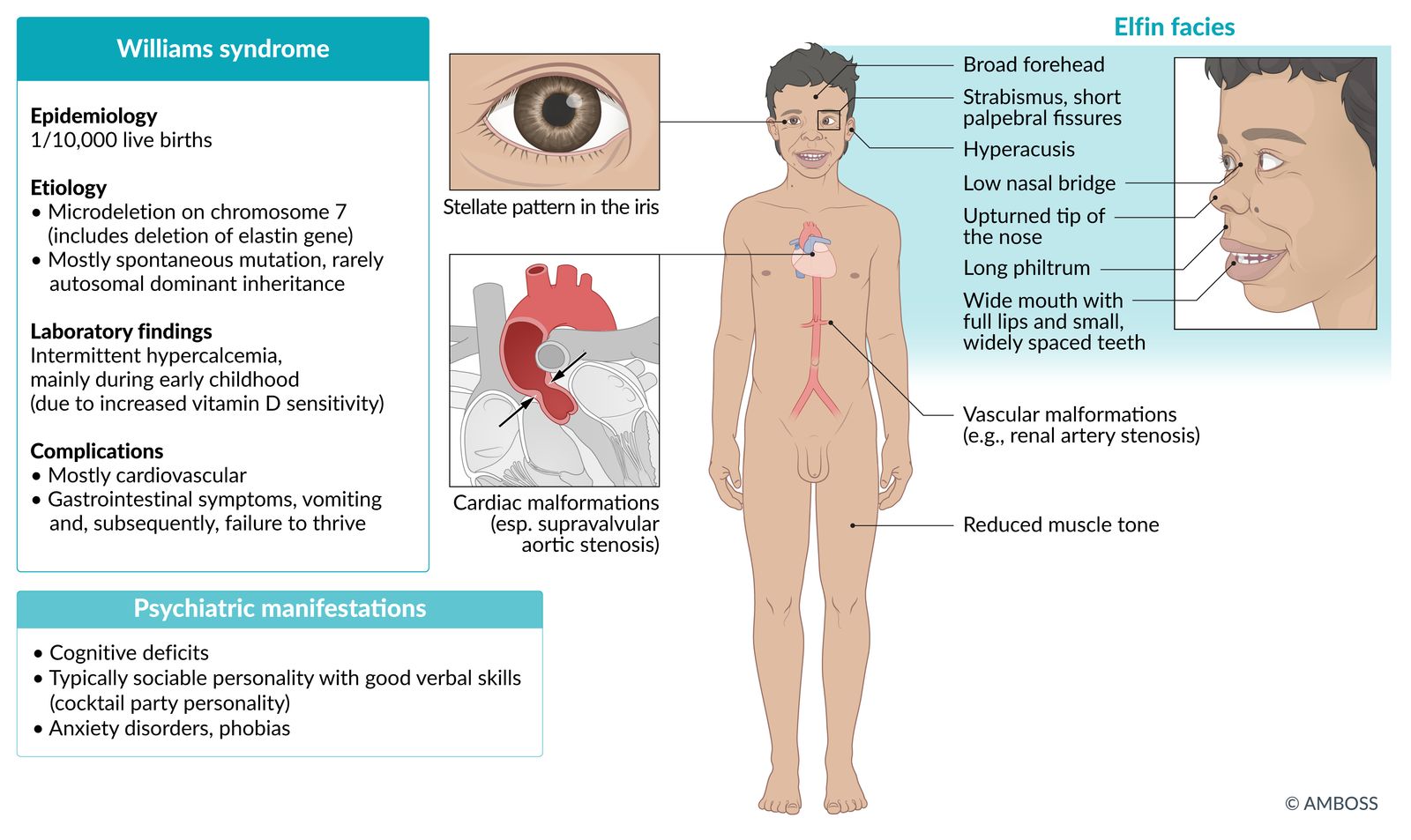
© AMBOSS
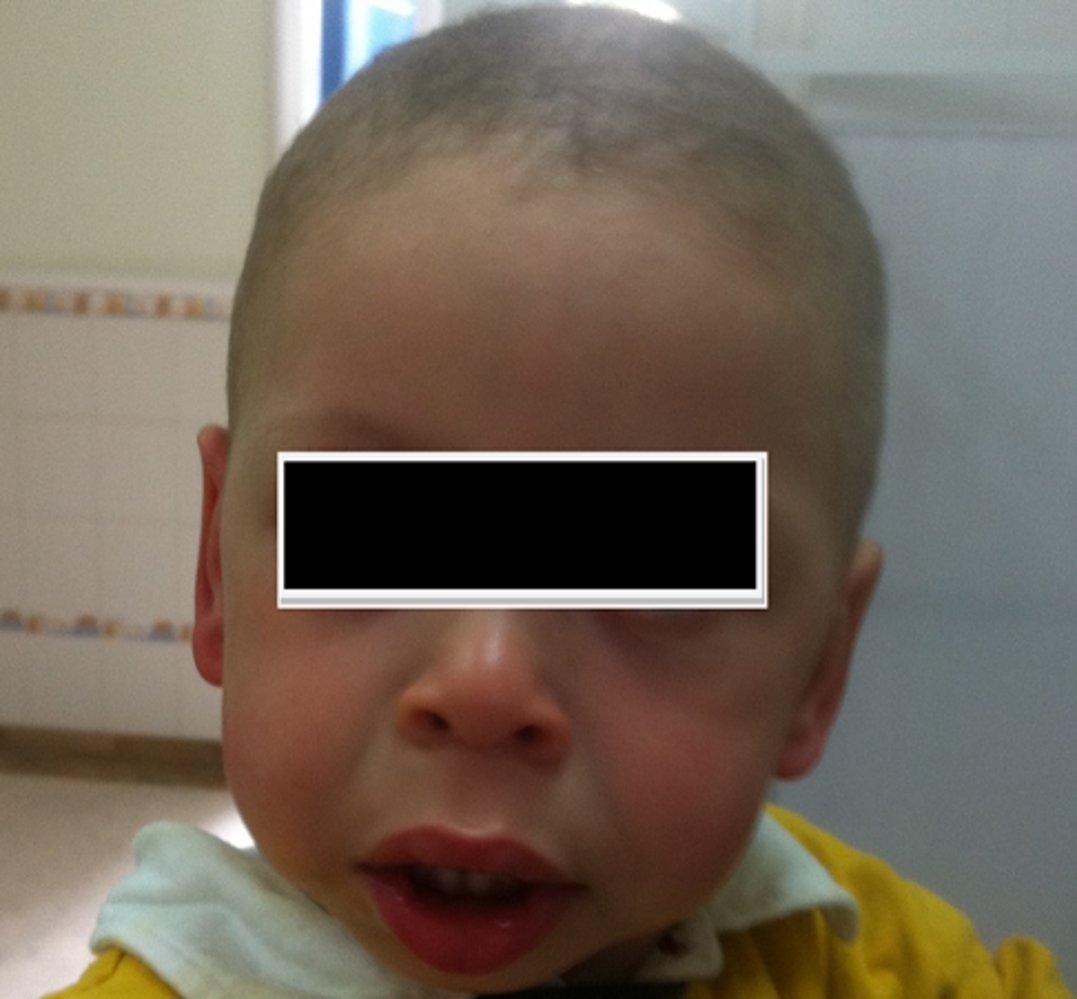
Photograph of a child with a broad forehead, midfacial hypoplasia, a short nose (with a flat nasal bridge), an everted lower lip, full cheeks and lips and low-set ears.
This appearance is often described as “elfin” facies and commonly seen in Williams syndrome, although definitive diagnosis would require genetic testing.
Source: “Figure 1. in: Williams-Beuren syndrome associated with single kidney and nephrocalcinosis: a case report” by Kamel Abidi, Manel Jellouli, Rania Ben Rabeh, Yousra Hammi, Tahar Gargah, The Pan African Medical Journal, licensed under CC BY 2.0.

Original title: “Genetics: Section 12.2 - Williams Syndrome”. Created by: Physeo™.
Zellweger syndrome (cerebrohepatorenal syndrome)
- Definition: an autosomal recessive defect in the PEX gene that results in impaired peroxisome synthesis [13][14]
-
Clinical features
- Characterized by presentation in the neonatal period with neonatal seizures and hypotonia
- Malformation of the face and head
- Hepatomegaly
- Prolonged jaundice and feeding difficulties
- Polycystic kidney disease (PKD)
- Retinal dystrophy and sensorineural hearing loss
- Diagnosis: increased concentration of very long chain fatty acids in the blood
-
Treatment
- Currently, there is no cure for Zellweger syndrome available.
- Supportive management, including:
- Placement of a feeding tube
- Consulting specialists to treat individual problems (e.g., audiologists, ophthalmologists, orthopedists)
- Prognosis: death within one year of birth
Pierre Robin sequence (Pierre Robin syndrome)
- Definition: : a set of abnormalities causing fetal oral and maxillofacial malformations [15]
-
Clinical features
- Cleft palate
- Glossoptosis (retraction of the tongue in the pharynx) with possible complications such as acute respiratory distress and aspiration
- Mandibular retrognathia (posterior mandibular positioning) or micrognathia (small lower jaw)
- Possible intellectual disability [16]
- Diagnosis: fluorescence in situ hybridization (FISH)
-
Treatment
-
If moderate dyspnea: symptomatic treatment
- Noninvasive ventilation
- Supervision and assistance while eating
-
If severe dyspnea: surgical correction
- Special interventions for long-term correction (e.g., mandibular distraction to position the tongue closer to the front of the mouth)
- In cases of acute life-threatening respiratory distress → tracheostomy
-
If moderate dyspnea: symptomatic treatment
Rubinstein-Taybi syndrome
- Definition: inherited syndrome with characteristic facial dysmorphia
- Etiology: CREBBP gene mutation
-
Clinical features
- Typical facial shape: highly arched eyebrows, beaked nose with hypoplastic wing of the nose
- During infancy: hairy forehead
- Short stature
- Broad thumbs and toes
- Intellectual disability
-
Prognosis
- Usually poor
- Infants born with this disorder usually survive only to early childhood
References:[17]
Smith-Lemli-Opitz syndrome
- Definition: autosomal recessive disease with cholesterol shortage due to deficient 7-dehydrocholesterol reductase [18][19]
- Etiology: gene mutation of DHCR7 (7-dehydrocholesterol reductase) at chromosome 11
-
Clinical features: Symptoms may vary widely.
- Short stature
- Dysmorphic facial features (ptosis, epicanthal folds, microcephaly, micrognathia)
- Intellectual disability
- Internal organs malformations (genitals, kidneys, heart)
- Toe syndactyly
- Single palmar crease
- Seizures
- Diagnosis: ↑ 7-dehydrocholesterol, ↓ cholesterol
- Treatment: cholesterol substitution
Albright hereditary osteodystrophy (Martin-Albright syndrome)
- Definition: a constellation of physical findings that occur in patients with pseudohypoparathyroidism type 1a and pseudopseudohypoparathyroidism
- Mode of inheritance: : usually autosomal dominant
-
Clinical features
- Short stature, round face, as well as metacarpal and metatarsal shortening (shortened fourth and fifth digits)
- Intellectual disability (oligophrenia)
- Additionally signs of hypoparathyroidism: hypocalcemia, tetany, cerebral seizures, intracranial calcifications, and tooth anomalies
-
Laboratory findings
- Pseudohypoparathyroidism type 1a: ↑ PTH, ↓ calcitriol, ↓ calcium, ↑ phosphate
- Pseudopseudohypoparathyroidism: normal PTH, calcitriol, calcium, and phosphate
Noonan syndrome
- Definition: autosomal dominant condition with a heterogeneous clinical picture due to mutation in the PTPN11 gene on chromosome 12 [20]
- Epidemiology: 1:1000–2500 live births
-
Clinical features
- Proportionate short stature (often developing only after birth)
- Minor facial dysmorphism, including ocular hypertelorism (a distance between the eyes that is greater than the 95th percentile); and downslanting eyes; webbed neck
- Heart disease (including pulmonary valve stenosis)
- Intellectual development may be delayed, but by adulthood intelligence is normal in ⅔ of patients.
-
Differential diagnoses
- Bardet-Biedl syndrome: a rare monogenic hereditary disease characterized by short stature, obesity, retinitis pigmentosa, intellectual disability, and polydactyly
- Weill-Marchesani syndrome: a rare hereditary disorder characterized by dwarfism, heart defects, and eye conditions (lenticonus, ectopia lentis, and secondary glaucoma)
Russell-Silver syndrome (Silver-Russell syndrome)
- Definition: rare, sporadic syndrome associated with intrauterine growth retardation
-
Clinical features
- ↓ Infant length
- Relative macrocephaly
- Dysmorphic facial features: asymmetric triangular face with a high forehead and drooping labial commissures
- Clinodactyly (crooked finger)
- Normal or mildly impaired cognitive development
Treacher Collins syndrome
- Definition: An autosomal dominant condition characterized by craniofacial abnormalities and external ear deformities resulting from impaired migration of neural crest cells into the first and second branchial arches. [21]
- Etiology: most commonly results from the mutations of TCOF1 gene on chromosome 5 or POLR1D gene on chromosome 5
-
Clinical features
- Micrognathia and retrognathia → difficulties with feeding and respiration
- Hypoplasia of the zygomatic arch → downward slant of the eyes
- External ear and auditory canal abnormalities → conductive hearing loss
- Coloboma of the lower lid
-
Management
- Respiratory support if needed
- Craniofacial reconstruction surgery
- Hearing aids
- Speech therapy
Pulmonary alveolar proteinosis (PAP)
| Overview of pulmonary alveolar proteinosis [22][23] | ||||
|---|---|---|---|---|
| Characteristics | Primary PAP | Secondary PAP | Congenital PAP | |
| Autoimmune PAP | Hereditary PAP | |||
| Definition |
|
|||
| Epidemiology [23] |
|
|
|
|
| Etiology |
|
|
|
|
| Pathophysiology |
|
|
|
|
| ||||
| Clinical features |
|
|||
| Diagnostics |
|
|||
| Treatment |
|
|
|
|
| Complications |
|
|||
Familial lipoprotein lipase deficiency
- Definition: an inherited genetic condition characterized by lipoprotein lipase (LPL) enzyme deficiency, which leads to hyperchylomicronemia
- Epidemiology: 1:250,000 [24]
- Etiology: : caused by mutations of the LPL gene
- Pathophysiology: gene mutations → LPL deficiency → ↓ breakdown of chylomicrons → hyperchylomicronemia
-
Clinical features
- Episodic epigastric pain of varying intensity
- Hepatosplenomegaly
- Eruptive cutaneous xanthomas
- Lipemia retinalis
- Neuropsychiatric symptoms (e.g., depression)
-
Diagnostics
- Clinical history
- Blood tests: ↓ LPL activity
- Confirmatory molecular genetic
- Treatment: lifestyle modifications (e.g., dietary restriction of fat, avoidance of alcohol and drugs that increase triglyceride levels) [25]
References:[24]
External Resources
References
- "Rubinstein-Taybi Syndrome". https://ghr.nlm.nih.gov/condition/rubinstein-taybi-syndrome. [2017-05-02]
- "Treacher Collins Syndrome". https://www.ncbi.nlm.nih.gov/pubmed/20301704. [1993-01-01]
- "Fragile X Syndrome". https://medlineplus.gov/ency/article/001668.htm. [2015-01-08]
- "Fragile X Syndrome". https://ghr.nlm.nih.gov/condition/fragile-x-syndrome. [2017-12-12]
- "Familial Lipoprotein Lipase Deficiency". https://rarediseases.org/rare-diseases/familial-lipoprotein-lipase-deficiency/
- Scott LJ. "Alipogene Tiparvovec: A Review of Its Use in Adults with Familial Lipoprotein Lipase Deficiency". Drugs. 75(2). :175-182. (2015)
- "Peroxisome Biogenesis Disorders, Zellweger Syndrome Spectrum". https://www.ncbi.nlm.nih.gov/books/NBK1448/. [2012-05-10]
- Karimi R, Brumfield T, Brumfield F, Safaiyan F, Stein S. "Zellweger syndrome: A genetic disorder that alters lipid biosynthesis and metabolism". The Internet Journal of Pharmacology. 5(1). (2006)
- "Rett Syndrome Information Page". https://www.ninds.nih.gov/Disorders/All-Disorders/Rett-Syndrome-Information-Page
- Mainardi PC. "Cri du Chat syndrome". Orphanet J Rare Dis. 1(33). (2006)
- Schiffer RB, Rao SM, Fogel BS. "Neuropsychiatry: A Comprehensive Textbook". LWW. (2003). ISBN: 0781726557
- Porter FD. "Smith–Lemli–Opitz syndrome: pathogenesis, diagnosis and management". Eur J Hum Genet. 16(5). :535-541. (2008)
- "Smith-Lemli-Opitz Syndrome"
- "Pulmonary Alveolar Proteinosis". https://rarediseases.org/rare-diseases/pulmonary-alveolar-proteinosis/
- McCarthy C, Avetisyan R, Carey BC, Chalk C, Trapnell BC. "Prevalence and healthcare burden of pulmonary alveolar proteinosis". Orphanet J Rare Dis. 13(1). (2018)
- "Prader-Willi Syndrome". https://rarediseases.org/rare-diseases/prader-willi-syndrome/. [2018-01-01]
- Mascari MJ, Gottlieb W, Rogan PK, et al. "The Frequency of Uniparental Disomy in Prader-Willi Syndrome". N Engl J Med. 326(24). :1599-1607. (1992)
- "Prader-Willi Syndrome". https://www.ncbi.nlm.nih.gov/books/NBK1330/. [2016-02-04]
- "Prader-Willi Syndrome". https://medlineplus.gov/ency/article/001605.htm. [2016-04-19]
- Robb MP. "Intro: A Guide to Communication Sciences and Disorders, Second Edition". Plural Publishing. (2013). ISBN: 9781597569279
- "Angelman Syndrome". https://www.ncbi.nlm.nih.gov/books/NBK1144/. [2015-05-14]
- "Angelman Syndrome". https://ghr.nlm.nih.gov/condition/angelman-syndrome. [2017-11-28]
- "Isolated Pierre Robin Sequence". https://ghr.nlm.nih.gov/condition/isolated-pierre-robin-sequence. [2017-05-02]
- Drescher F, Jotzo M, Goelz R, et al. "Cognitive and psychosocial development of children with Pierre Robin sequence". Acta Paediatr. 97(5). :653-656. (2008)
- Bhambhani V, Muenke M. "Noonan syndrome". Am Fam Physician. 89(1). :37-43. (2014)