Summary
Thalassemias are a group of hereditary hemoglobin disorders characterized by mutations on the α- or β-globin chains (resulting in alpha or beta thalassemia). Thalassemias can be further classified according to the specific genotype: the α-chain is coded by four alleles, resulting in four possible variants depending on the number of alleles affected, while the β-chain is coded by two alleles, resulting in two possible variants. The number of alleles affected is directly related to the severity of the disease (minor/intermedia/major). Thalassemia mutations are generally more frequent in areas where malaria is endemic; alpha thalassemias occur most commonly in individuals of Asian or African descent, whereas beta thalassemias are predominant in individuals of Mediterranean descent. The key feature in all forms of thalassemia is microcytic hypochromic anemia (which may be very mild in minor forms), but more severe forms may also manifest with hemolysis, splenomegaly, delay in growth and development, and skeletal deformities. The diagnostic workup for suspected thalassemia includes a blood smear, hemoglobin electrophoresis, high-performance liquid chromatography (HPLC), and, possibly, genetic testing. Minor forms of thalassemia usually require no treatment, while patients with thalassemia intermedia/major typically require regular blood transfusions and management of disease and treatment-related complications (e.g., chelating agent for transfusion-mediated iron overload).
Overview
| Overview of thalassemia | |||
|---|---|---|---|
| Type | Mutated gene | Clinical features | |
| Alpha thalassemia | Silent carrier |
|
|
| Alpha-thalassemia trait |
|
|
|
| Hemoglobin H disease |
|
|
|
| Hemoglobin Bart's disease |
|
|
|
| Beta thalassemia | Beta-thalassemia minor |
|
|
| Beta-thalassemia intermedia |
|
|
|
| Beta-thalassemia major |
|
|
|
| Sickle cell beta thalassemia |
|
|
|
Epidemiology
- Beta thalassemia: most commonly seen in people of Mediterranean descent
- Alpha thalassemia: most commonly seen in people of Asian and African descent
- Thalassemia provides partial resistance against malaria.
Alpha thalassemia is common in Asia and Africa.
Epidemiological data refers to the US, unless otherwise specified.
Etiology
General [1][2]
-
Cause: gene mutations
-
Beta thalassemia: usually due to point mutations in promoter sequences or splicing sites
- β-globin locus - short arm of chromosome 11
-
Alpha thalassemia: usually due to deletion of at least one out of the four existing alleles
- The α-globin gene cluster is located on chromosome 16
-
Beta thalassemia: usually due to point mutations in promoter sequences or splicing sites
- Inheritance pattern: autosomal recessive
Alpha thalassemia
In a normal cell, the α-globin chains are coded by a total of four alleles. Thus, there are four forms of the disease. The severity of alpha thalassemia depends on the number of defective α-globin alleles.
- Silent carrier (minima form): one defective allele (-α/αα)
-
Alpha-thalassemia trait (minor form)
- Two defective alleles (-α/-α or --/αα)
- Cis-deletion is common among Asian populations, whereas trans-deletions are more common in African populations.
- Children of parents with a two-gene deletion in cis are at higher risk of developing Hb Bart.
- Hemoglobin H disease (intermedia form): three defective alleles (--/-α) → results in excessive production of pathologically altered HbH
- Hemoglobin Bart's disease (major form): four defective alleles (--/-‑) → results in excessive production of pathologically altered Hb Bart (consists of four γ-chains; γ-tetramers)
Beta thalassemia [3][4]
In a normal cell, the β-globin chains are coded by a total of two alleles.; Thus, there are two main forms of the disease.
- Beta-thalassemia minor (trait): one defective allele
- Beta-thalassemia major (Cooley anemia): two defective alleles
- Sickle cell beta thalassemia: a combination of one defective β-globin allele and one defective HbS allele
- Hemoglobin E/beta thalassemia: a combination of one allele with a hemoglobin E (HbE) variant and one defective β-globin allele. Produces a highly heterogeneous clinical spectrum, and in severe cases patients present with features of beta-thalassemia major. [5]
- Hemoglobin E disease: a condition characterized by homozygosity to the HbE variant. Patients can present with mild features resembling beta-thalassemia minor (i.e., mild anemia).
Pathophysiology
Anemia results from a combination of inefficient erythropoiesis and increased hemolysis. The degree to which both mechanisms contribute to the severity of the disease depends on a patient's exact genotype. [6]
-
Inefficient erythropoiesis → anemia
-
Beta-thalassemia minor and major: faulty β-globin chain synthesis → ↓ β-chains→ ↑ γ-,δ-chains → ↑ HbF and ↑ HbA2.
- HbF protects infants up to the age of 6 months, after which HbF production declines and symptoms of anemia appear.
- Alpha-thalassemia intermedia (HbH disease) and alpha-thalassemia major (Bart's disease): faulty α-globin chain synthesis → ↓ α-chains → impaired pairing of α-chains with β-chains and γ-chains→ ↑ free β-, γ-chains → ↑ HbH, ↑ Hb-Bart's
- In minor and minima forms, production of the affected chain is reduced, but enough is produced to prevent severe anemia.
-
Beta-thalassemia minor and major: faulty β-globin chain synthesis → ↓ β-chains→ ↑ γ-,δ-chains → ↑ HbF and ↑ HbA2.
- Increased hemolysis: One of the chains (either α or β) is reduced → compensatory overproduction of other chains → excess globin chains precipitate and form inclusions within RBCs → erythrocyte instability with hemolysis
- Anemia → ↑ erythropoietin → bone marrow hyperplasia and skeletal deformities
Clinical features
Beta thalassemia
-
Minor variant
- No or mild anemia
- Low risk of hemolysis or splenomegaly
-
Major variant
- Severe hemolytic anemia that often requires transfusions → secondary iron overload due to hemolysis, transfusion, or both → secondary hemochromatosis [7]
- Hepatosplenomegaly
- Growth retardation
- Skeletal deformities (high forehead, prominent zygomatic bones, and maxilla)
- Transient aplastic crisis (secondary to infection with parvovirus B19) [8]
-
Sickle cell beta thalassemia
- Features of sickle cell disease
- Severity depends on the amount of β-globin synthesis.
Alpha thalassemia
- Silent carrier: typically no anemia
- Alpha-thalassemia trait: : no or mild anemia
-
Hemoglobin H disease
- Jaundice and anemia at birth
- Chronic hemolytic anemia that may require transfusions → secondary iron overload due to hemolysis, transfusion, or both → secondary hemochromatosis [7]
- Hepatosplenomegaly
- Skeletal deformities (less common)
- Compared to thalassemia beta, symptoms in adults are generally less severe.
-
Hb-Bart's hydrops fetalis syndrome (most severe variant of alpha thalassemia)
- Intrauterine ascites and hydrops fetalis
- Severe hepatosplenomegaly
- Often cardiac and skeletal anomalies
- Incompatible with life (death in utero or shortly after birth)
Diagnosis
Pretest probability [9][10]
The presentation of thalassemia is highly variable, ranging from incidental findings to life-threatening forms. Thalassemia is more likely to be diagnosed in patients with the following:
-
Suggestive clinical features
- In infants (usually aged 6–24 months): Consider beta-thalassemia major.
- In children (usually aged 2–6 years): Consider beta-thalassemia minor or alpha/beta-thalassemia intermedia.
-
Demographic factors
- Family history of thalassemia
- Asian, African, or Mediterranean ancestry
-
Incidental diagnostic findings
- Microcytic hypochromic anemia not explained by other causes or not responsive to supplemental iron
- Antenatal ultrasound showing hydrops fetalis
- Detection of abnormal hemoglobin chains in newborn screening [9]
Family history plays an important role in diagnosing patients with clinically silent thalassemia. Consider the possibility of minor forms/traits if a family member is diagnosed with a more severe form.
Initial investigations [9][10]
-
CBC
- Characteristic finding: microcytic hypochromic anemia (i.e., MCV < 80 fL, MCH < 27 pg/cell) present regardless of Hb level
- Hb levels: variable depending on the subtype
- Other red cell indices
- Normal RDW [11]
- Higher RBC count than iron deficiency anemia
-
Mentzer index
- MCV/RBC ratio used to differentiate between thalassemia and iron deficiency anemia (IDA)
- A ratio < 13 suggests thalassemia; a ratio > 13 suggests IDA
- See also “Diagnostics of anemia” for further evaluation of microcytosis.
-
Hemolysis evaluation: nonimmune-mediated hemolytic anemia
- ↓ Haptoglobin, ↑ LDH, ↑ reticulocytes
- Liver chemistries: hyperbilirubinemia (indirect)
- Coombs test: negative
- Iron studies (particularly ferritin) : expected to be normal in thalassemia
-
Peripheral blood smear findings include:
- Target cells
- Teardrop cells
- Anisopoikilocytosis
- HbH inclusion bodies
- Erythroblasts
")

Confirmatory diagnostic studies [9][10]
-
Detection of hemoglobin variants
- Hb-electrophoresis (qualitative analysis)
- Automated HPLC (qualitative and quantitative analysis)
-
Findings (vary depending on the subtype)
- Hemoglobin A (and subtypes): hemoglobin A2 values are helpful to determine the diagnosis (i.e., to distinguish alpha from beta thalassemia)
- Hemoglobin F: may be elevated in some children and adults with thalassemia
- See “Hemoglobin variants” for findings of other hemoglobinopathies.
| Interpretation of Hb-electrophoresis results for thalassemia [10][12][13] | |||
|---|---|---|---|
| Alpha thalassemia | Beta-thalassemia minor/intermedia/major | ||
| Minor | Intermedia/HbH disease | ||
| MCV/MCH | Normal/low | Low | Low |
| HbA2 | Normal/low | Normal/low | High |
| HbF | Normal | Normal/high | High |
| HbH | May be present | Present | Absent |
- Genetic studies: (PCR-based): to determine specific diagnosis and mutations
-
Bone marrow aspiration (not routinely indicated)
- Usually performed to rule out other hematologic conditions
- Findings in thalassemia are nonspecific (e.g., reactive hyperplasia).
Beta-thalassemia minor should be strongly suspected if HbA2 is > 3.5%. [12]
Imaging
Imaging is not routinely indicated or required for diagnosis. It can be useful in the evaluation of suspected craniofacial abnormalities and extramedullary hematopoiesis.
-
Skull x-ray (AP and lateral)
- Indication: assessment of craniofacial abnormalities
- Findings include:
- High forehead
- Prominent zygomatic bones and maxilla (referred to as “chipmunk facies”)
- Hair-on-end (also known as “crew cut”) sign
-
CXR
- Indication: suspected extramedullary hematopoiesis in the thorax
- Findings include:
- Mediastinal or pulmonary masses
- Subperiosteal extension in the ribs (also known as “rib within a rib”)
- MRI spine: helpful to evaluate mass effect symptoms due to extramedullary hematopoietic pseudotumors



CBC parameters can help differentiate thalassemia minor/trait from iron deficiency anemia (IDA), which is frequently associated with a high RDW, low RBC count, and low MCV, typically occurring once Hb is < 10 g/dL. In thalassemia, microcytosis is always present regardless of Hb level, and RDW is normal to increased. Also, RBC count is higher and MCV is lower than in IDA.
Low ferritin suggests iron deficiency anemia and patients should receive iron supplementation. Suspect thalassemia if there is no significant response after three months. [9][10]
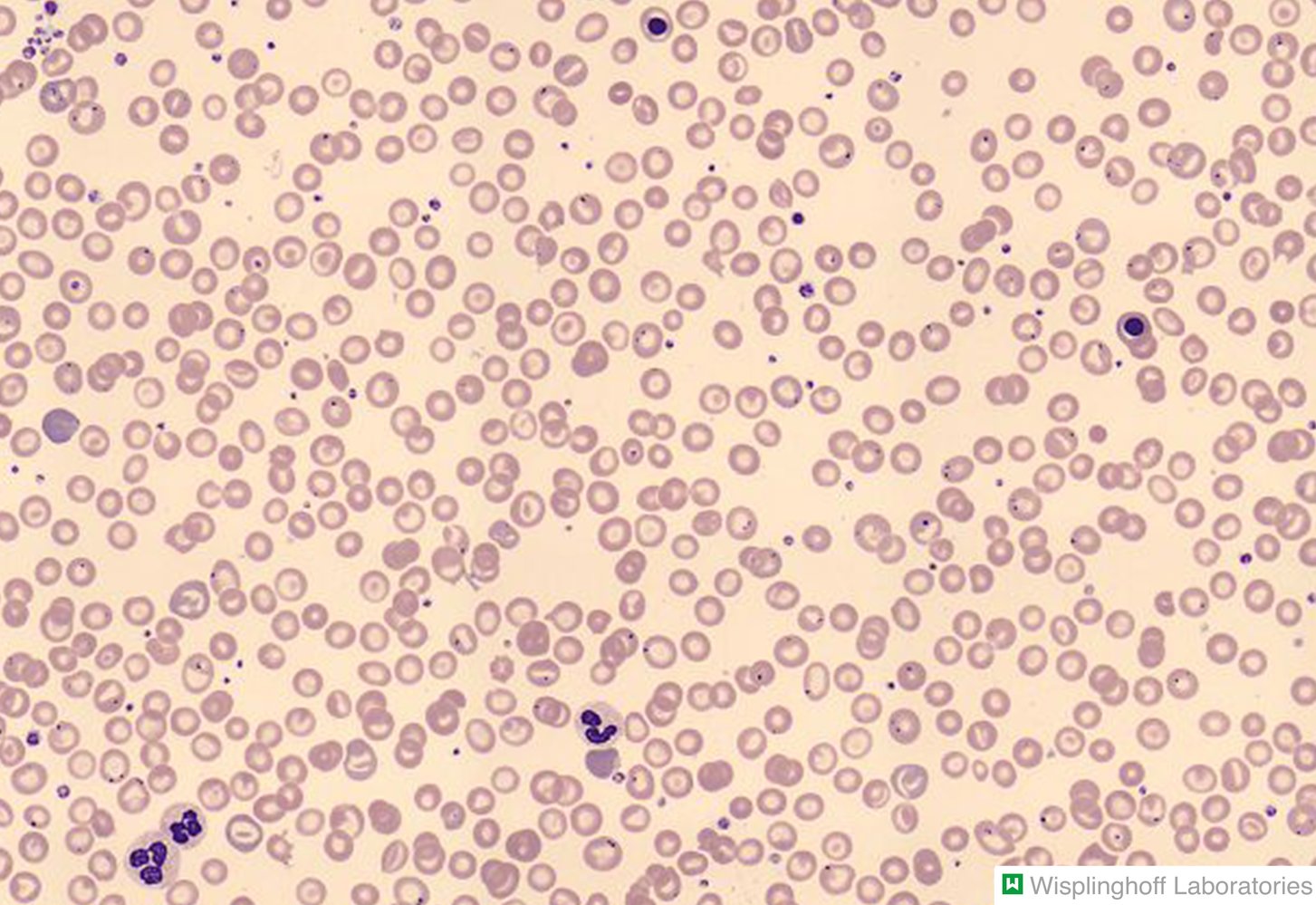
Photomicrograph of a peripheral blood smear
Hypochromic erythrocytes with a dark central area, a pale ring surrounding the center, and a dark outer rim can be seen (dashed red circles on overlay). Due to this “bullseye” appearance on light microscopy, they are called target cells. On electron microscopy, they would appear bell-shaped (overlay illustration; visual axis in light microscopy depicted in red).
The finding of target cells is nonspecific and may indicate various hemoglobinopathies, such as thalassemia, liver disease, or asplenia.
Our great thanks to Dr. Wisplinghoff (Dr. Wisplinghoff's laboratory) for kindly providing this image.
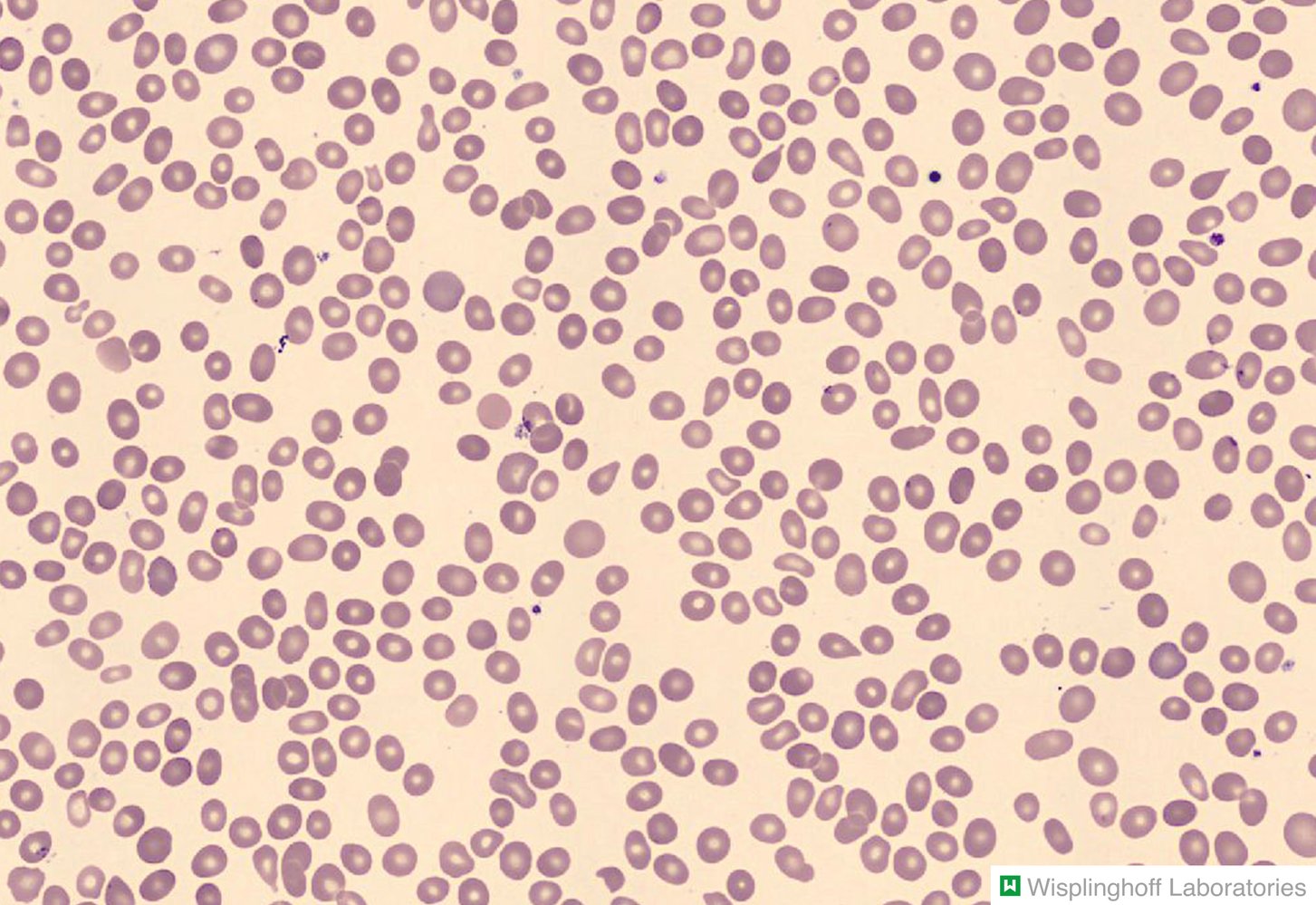
Photomicrograph of a peripheral blood smear
Erythrocytes with a teardrop shape can be seen throughout the image (red overlay). The majority of the erythrocytes appear round and normal.
Teardrop-shaped erythrocytes, known as dacrocytes, are found in conditions that involve extramedullary hematopoiesis (e.g., myelofibrosis, thalassemia, splenomegaly).
Our great thanks to Dr. Wisplinghoff (Dr. Wisplinghoff's laboratory) for kindly providing this image.
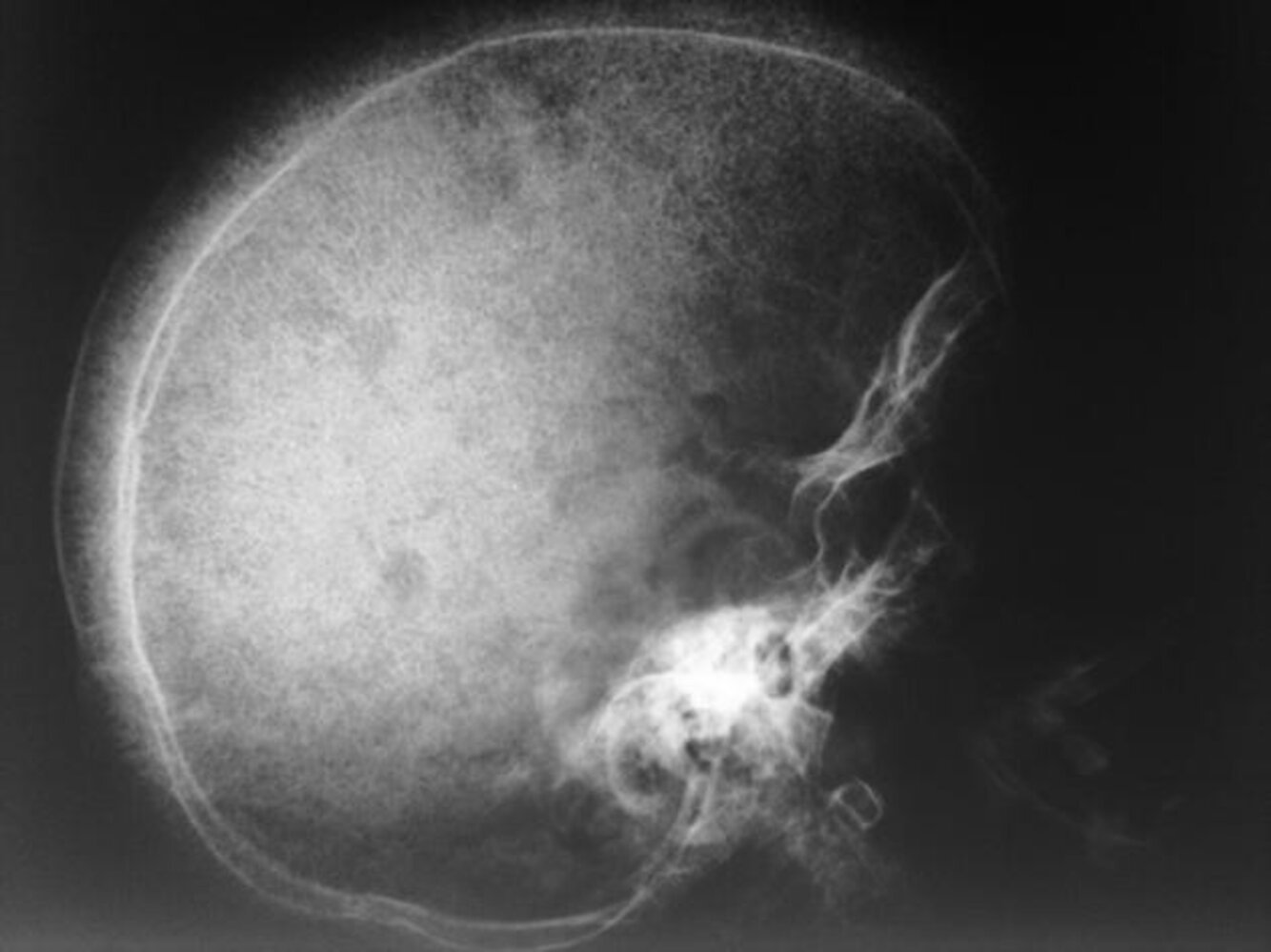
X-ray skull (lateral view) of a child with beta thalassemia
Expansion of the diploic space with thickening of the trabeculae (example indicated by green overlay) is the result of extramedullary hematopoiesis. The hair-on-end appearance (examples indicated by red lines) produced can also be seen in other chronic hemolytic anemias.
Source: “WBR0150” by W. J. Gibson, WikiDoc, licensed under CC BY-SA 3.0. The supplementary image with overlays of relevant areas was adapted from the image mentioned above and licensed under CC BY-SA 3.0.
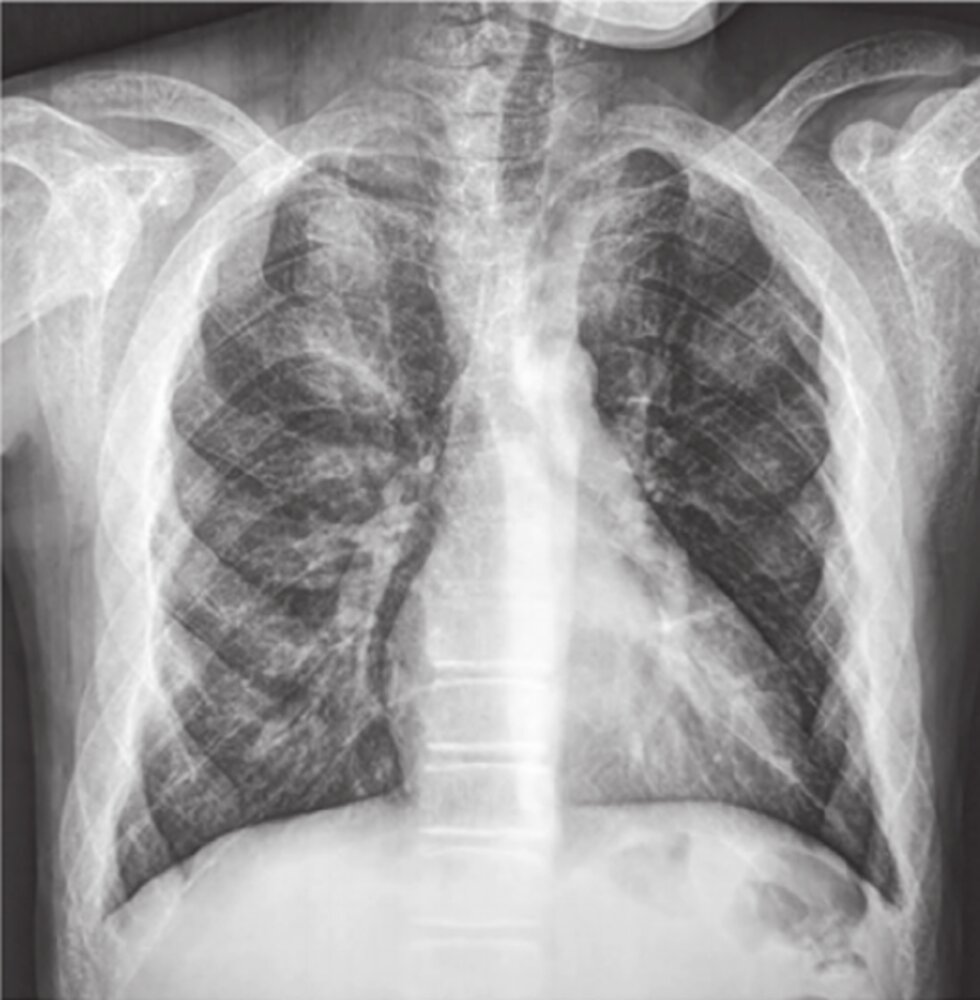
X-ray chest (PA view) of a patient with beta-thalassemia major
Skeletal structures show decreased density and a coarsened heterogeneous appearance. The ribs are expanded and additional lines of increased density parallel the outer edges of the middle and anterior aspects of several ribs are visible (examples of double lines indicated by green lines). The appearance is the result of subperiosteal marrow proliferation and has been termed the “rib-within-a-rib sign.”
Source: “Fig 1c, In: Spinal cord compression by extramedullary hematopoiesis in beta-thalassemia major” by Ammar LB, Ferjani H, Maatallah K et al., Wiley Online Library, licensed under CC BY 4.0. Modifications: letter removed. The supplementary image with overlays of relevant areas was adapted from the image mentioned above (© AMBOSS).
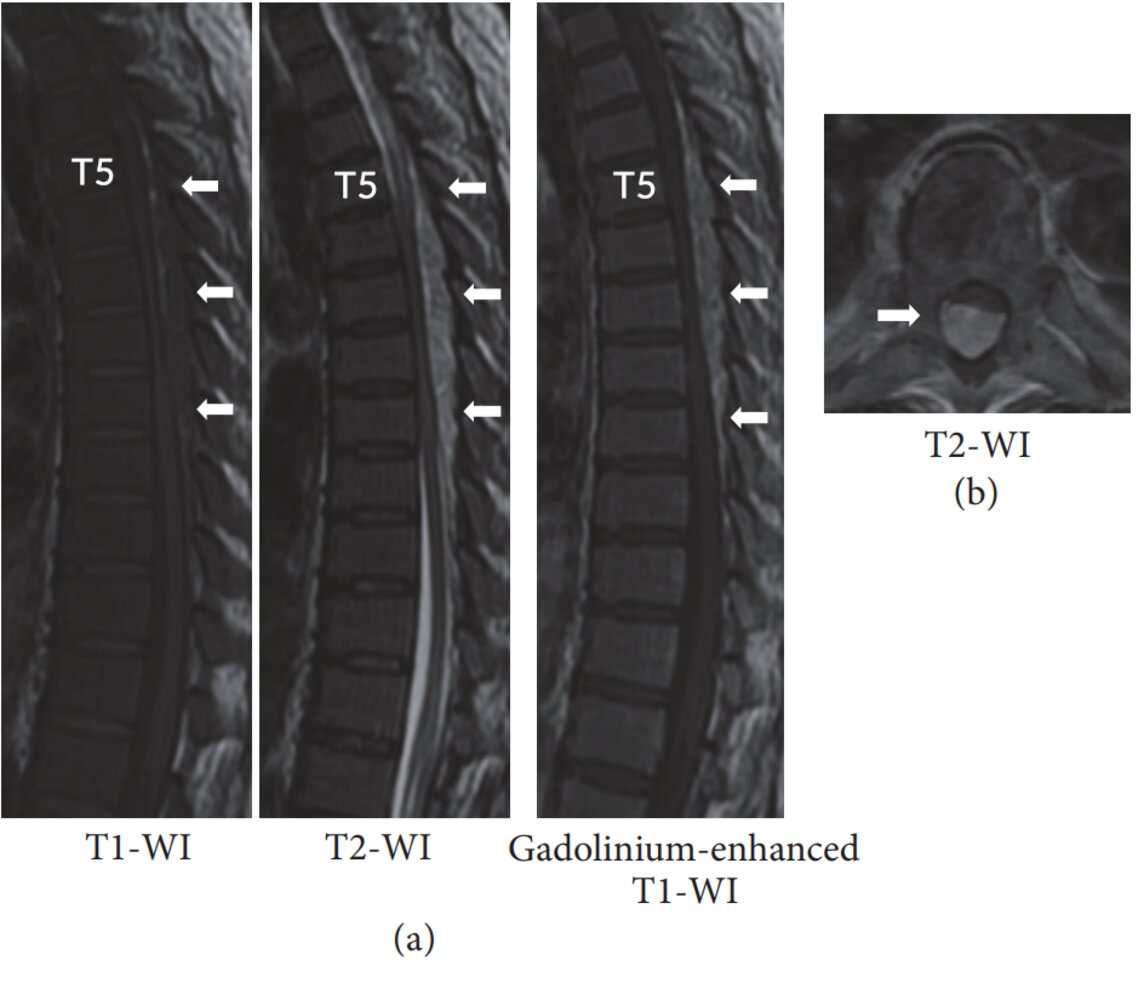
MRI thoracic spine (a: T1-weighted, T2-weighted, and T1-weighted gadolinium-enhanced; sagittal plane; b: T2-weighted; axial plane) of a patient with polycythemia vera
A posterior epidural mass extends from T5–T10, narrowing the spinal canal. The lesion shows intermediate signal intensity on T1-weighted images, slightly high signal intensity on T2-weighted images, and mild gadolinium enhancement.
The MRI features suggest an active lesion of extramedullary hematopoiesis. The signal intensity of extramedullary hematopoiesis can vary with lesion activity and amount of hematopoietic tissue.
Source: “Figure 1 in: Myelopathy due to Spinal Extramedullary Hematopoiesis in a Patient with Polycythemia Vera” by Shuhei Ito et al., Case Reports in Orthopedics, licensed under CC BY 4.0. Modifications: Changed from Th5 to T5.
Management
-
All patients
- Patient education and genetic counseling.
- Screening tests for relatives
-
Thalassemia minor
- Usually no treatment required
- Episodic folic acid supplementation may be indicated (e.g., during pregnancy, acute infections)
-
Thalassemia major and intermedia
-
Transfusion therapy (erythrocyte concentrates)
- Indication (for transfusion-dependent thalassemias): Hb < 7 g/dL or marked clinical symptoms
- Target: Hb > 9–10 g/dL
- Surveillance and treatment of complications
- Iron overload diseases: chelating agents, e.g., deferasirox, indicated when iron accumulation reaches toxic levels
- Other complications: e.g., gallstones, asplenia, extramedullary hematopoietic pseudotumors
-
Transfusion therapy (erythrocyte concentrates)
-
Select patients
- Splenectomy
- Potentially curative treatment
- Stem cell transplantation: allogenic HSCT
- Gene therapy
References: [9][10]
Anemia
Transfusion therapy [9][10][14]
This is the mainstay of management for thalassemia major and intermedia (see “Transfusion” for more information about pretransfusion testing and transfusion reactions).
- Transfusion dependency: can fluctuate for individual patients depending on the subtype, severity, and external factors.
- Non-transfusion-dependent patients: only require either occasional or short-term regular blood transfusions for acute needs.
- Transfusion-dependent patients: require lifelong regular transfusions (e.g., every 2–5 weeks).
| Transfusion therapy in thalassemias | ||
|---|---|---|
| Non-transfusion-dependent thalassemias (NTDT) [9] | Transfusion-dependent thalassemias [10] | |
| Subtypes |
|
|
| Indications for transfusion |
|
|
| Goals of therapy |
|
|
Additional therapies
-
Folic acid should be considered in patients with: [14]
- Thalassemia major or intermedia: regular supplementation
- Thalassemia minor during periods of acute physiological stress (e.g., infections): episodic supplementation
- Fetal hemoglobin induction: hydroxyurea may help induce fetal hemoglobin, reducing symptoms and the need for transfusions
Splenectomy [9][10]
- Limited use: risks may outweigh benefits (see “Asplenia”).
-
Indications include:
- Hypersplenism causing recurrent infections or bleeding
- Clinically significant splenomegaly
- Symptomatic (e.g., abdominal pain)
- Massive splenomegaly (> 20 cm) at risk of splenic rupture
- Uncontrollable iron overload disease
- Uncontrollable anemia affecting growth and development
- Post-operative care: See “Management of asplenic patients.”
Avoid splenectomy in patients < 5 years old due to the risk of overwhelming postsplenectomy sepsis.
Iron overload disease
All patients receiving transfusion therapy should be periodically evaluated for iron overload disease and subsequent organ damage. [9][10][12]
- Clinical features of iron overload diseases: e.g., bronze skin, growth delay, signs of organ damage
-
Diagnosis of iron overload diseases
- Liver biopsy (gold standard test)
- MRI (noninvasive alternate test)
- General monitoring: serum ferritin
-
Monitoring for organ damage
- Endocrinopathies: Screen patients for the following conditions and refer to endocrinology as needed.
- DM
- Hypothyroidism/hypoparathyroidism
- Hypogonadism
- Osteoporosis
- Liver cirrhosis and hepatocarcinoma: liver chemistries (every 3 months) and MRI (annually)
- Cardiac siderosis: regular imaging (echocardiogram or MRI) [10][15]
- Endocrinopathies: Screen patients for the following conditions and refer to endocrinology as needed.
-
Treatment: Chelation therapy is typically recommended when iron accumulates to toxic levels and may be required from a very early age. [9][10][12]
- Deferasirox (first-line)
- Deferoxamine
- Deferiprone
Iron overload can seriously affect the liver and cardiac function, as well as several endocrine glands, and is potentially lethal in the long term. Once organ damage occurs, it is often irreversible.
The objective of chelation therapy is to prevent organ damage resulting from iron overload disease and requires good adherence to treatment, continuous monitoring by specialists, and frequent dosing adjustment.
Other chronic complications
In addition to iron overload disease, patients may develop other long-term complications secondary to the disease or its treatment.
| Common complications in patients with thalassemia [9][10][12] | |||
|---|---|---|---|
| Mechanism | Management | ||
| Hepatobiliary complications | Cholelithiasis |
|
|
| Liver disease |
|
|
|
| Hematologic complications | Hypercoagulable states |
|
|
| Hemolytic crisis |
|
|
|
| Extramedullary hematopoietic pseudotumors |
|
|
|
| Cardiovascular complications |
|
|
|
| Chronic leg ulcers |
|
|
|
| Mental health complications |
|
|
|
Hematopoietic stem cell transplant (HSCT)
HSCT can have good outcomes and be considered curative, however, its use is limited due to high mortality and morbidity. Specialist evaluation and shared decision-making (involving patients and/or surrogate decision-makers) are essential and should weigh each patient's individual risks and benefits. [10]
-
Modalities
- Compatible sibling donor (preferred): most successful alternative; mortality rate of ∼ 5%
- Matched unrelated donor (alternative): can be considered; higher chances of rejection
-
Limitations
- Requires the availability of a compatible donor and access to an HSCT specialized center
- Must be performed during early childhood, before iron overload is present (which decreases success) [10]
- Aggressive pretransplant myeloablation → ↑ transfusion requirement → ↑ risk of iron overload disease
- Posttransplant immunosuppression → ↑ infection risk
Special patient groups
Thalassemia in pregnancy [16][17][18][19]
-
Overview
- Most patients with thalassemia may suffer from infertility and require assisted reproductive technology to achieve pregnancy.
- Spontaneous fertility may occur in patients who have successfully undergone iron chelation therapy and blood transfusion. [20]
-
Pregnancies in patients with thalassemia should be planned, as they are considered high-risk for both the mother and fetus.
- Preconception genetic counseling, close antenatal monitoring and screening, and intrapartum and postpartum management are highly recommended.
- A multidisciplinary team, including a hematologist, cardiologist, endocrinologist, and gynecologist, should be involved in the management of pregnant patients with thalassemia.
Fertility assessment and management
-
Screening for thalassemia
- Hemoglobinopathy screening should be offered to women with unknown hemoglobinopathy status and normocytic or microcytic anemia.
-
Partner screening for thalassemia carrier status
- Not a carrier for thalassemia: Follow protocols for spontaneous conception, ovulation induction (e.g., gonadotropins, clomiphene citrate), or in vitro fertilization.
- Carrier of alpha thalassemia: Offer preimplantation genetic testing and in vitro fertilization.
- Partner has thalassemia trait: Advise patients to use donor gametes or a sperm donor.
- If both partners have thalassemia or thalassemia trait: Offer genetic counseling prior to conception. After successful conception, the fetus will undergo thalassemia screening using chorionic villus sampling or amniocentesis.
- If both partners have thalassemia major: Consider alternatives including adoption.
-
Preconception screening and review: The most important factors to consider during a fertility assessment are cardiac and liver function, and risk of vertical transmission of viral diseases. Diagnostics include:
- Ovarian reserve: anti-Müllerian hormone levels
- CBC (MCH, MCV) and RBC indices
- Hb electrophoresis
- Iron studies
- Cardiac function: echocardiogram, electrocardiogram, T2 star cardiac MRI
- Liver function: liver function tests, liver elastography, liver and gallbladder ultrasound
- Bone density: DEXA scan, vitamin D, calcium levels
- Endocrine function: thyroid function tests, blood glucose levels
- Coagulation studies: PT, INR, protein C, protein S, homocysteine, thrombophilia panel
- Viral infection screening: HBV, HCV, HIV
- Screening for RBC antibodies
- Medication review: folic acid, vitamin D, calcium supplementation, oral iron chelators
Antepartum screening
-
Evaluation
- Cardiac function tests each trimester
- Gestational diabetes screening at 16 weeks' gestation
- Fetal growth monitoring: serial fetal biometric ultrasounds starting from 24 weeks' gestation
- Assessment of splenomegaly
-
Iron chelation treatment: should be adjusted and closely monitored during pregnancy
- Deferoxamine: used in the second and third trimesters
- Deferasirox and deferiprone: must be discontinued 3 months prior to conception
- Transfusions: should aim to maintain hemoglobin > 10 g/dL to prevent intrauterine fetal growth restriction
Intrapartum and postpartum management
- Although vaginal delivery is possible, patients usually require cesarean delivery, as those with thalassemia have an increased incidence of cephalopelvic disproportion.
- Breastfeeding should not be discouraged.
- Resume iron chelation therapy, if eligible.
- Deferoxamine can be resumed immediately postpartum.
- The use of deferasirox and deferiprone should be delayed until the patient has stopped breastfeeding.
- Continue calcium and vitamin D supplementation.
- Thromboprophylaxis with LMW heparin
- Discuss contraception options (i.e., progesterone-only pill or barrier methods).
Complications
-
Maternal
- Cardiac failure
- Alloimmunization
- Thrombosis
- Diabetes mellitus
- Hypothyroidism
- Hyperparathyroidism
-
Fetal
- Growth restriction
- Prematurity
- Transmission of hepatitis B, hepatitis C, and/or HIV
Related One-Minute Telegram
- One-Minute Telegram 17-2021-1/3: A CRISPR-Cas miracle? Report on two successful attempts at treatment
Interested in the newest medical research, distilled down to just one minute? Sign up for the One-Minute Telegram in “Tips and links” below.
External Resources
References
- Singh AK, Loscalzo J. "The Brigham Intensive Review of Internal Medicine". Oxford University Press, USA. (2014). ISBN: 9780199358274
- Benjamin I. "Andreoli and Carpenter's Cecil Essentials of Medicine". Elsevier Health Sciences. (2016). ISBN: 9781437718997
- Fibach E, Rachmilewitz EA. "Pathophysiology and treatment of patients with beta-thalassemia – an update". F1000Res. 6. :2156. (2017)
- Galanello R, Origa R. "Beta-thalassemia". Orphanet J Rare Dis. 5. :11. (2010)
- Fucharoen S, Weatherall DJ. "The Hemoglobin E Thalassemias". Cold Spring Harb. Perspect. Med.. 2(8). :a011734-a011734. (2012)
- Rivella S. "Ineffective erythropoiesis and thalassemias". Curr Opin Hematol. 16(3). :187-194. (2009)
- "Secondary Iron Overload". https://www.merckmanuals.com/professional/hematology-and-oncology/iron-overload/secondary-iron-overload. [2017-01-01]
- Arabzadeh SA, Alizadeh F, Tavakoli A, et al. "Human parvovirus B19 in patients with beta thalassemia major from Tehran, Iran.". Blood research. 52(1). :50-54. (2017)
- "Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT)". https://www.ncbi.nlm.nih.gov/books/NBK190453/. [2017-01-01]
- "Guidelines for the Management of Transfusion Dependent Thalassaemia (TDT)". https://www.ncbi.nlm.nih.gov/pubmed/25610943. [2014-01-01]
- Van Vranken M. "Evaluation of microcytosis.". Am Fam Physician. 82(9). :1117-22. (2010)
- Caligiuri M, Levi MM, Kaushansky K, et al. "Williams Hematology, 9E". McGraw-Hill Education / Medical. (2015). ISBN: 9780071833004
- Mohan H. "Textbook of Pathology 8th edition". JP Medical Ltd. (2018). ISBN: 9789352705474
- Rachmilewitz EA, Giardina PJ. "How I treat thalassemia". Blood. 118(13). :3479-3488. (2011)
- Pennell DJ, Udelson JE, Arai AE, et al. "Cardiovascular Function and Treatment in β-Thalassemia Major". Circulation. 128(3). :281-308. (2013)
- Tsironi M, Petrakos G, Andriopoulos P. "Pregnancy in women with thalassemia: challenges and solutions". International Journal of Women's Health. Volume 8. :441-451. (2016)
- "2021 GUIDELINES FOR THE MANAGEMENT OF TRANSFUSION DEPENDENT THALASSAEMIA (TDT)". https://www.thalassemia.org/boduw/wp-content/uploads/2021/06/TIF-2021-Guidelines-for-Mgmt-of-TDT.pdf. [2021-01-01]
- Taher A, Vichinsky EP, Federation TI, et al. "Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT)". Thalassaemia International Federation. (2013). ISBN: 9789963717033
- Origa R, Comitini F. "PREGNANCY IN THALASSEMIA". Mediterranean Journal of Hematology and Infectious Diseases. 11(1). :e2019019. (2019)
- Singer ST, Vichinsky EP, Gildengorin G, et al. "Reproductive capacity in iron overloaded women with thalassemia major". Blood. 118(10). :2878-2881. (2011)
- "Thalassemia". https://medlineplus.gov/ency/article/000587.htm. [2017-03-09]
- "Thalassemia". http://www.mayoclinic.org/diseases-conditions/thalassemia/symptoms-causes/dxc-20261829. [2016-11-02]
- "Blood smear". https://medlineplus.gov/ency/article/003665.htm. [2017-03-09]