Summary
Von Willebrand disease (vWD) is a bleeding disorder characterized by a deficiency or dysfunction of von Willebrand factor (vWF). vWD is the most common congenital bleeding disorder, affecting approximately 1% of the US population. vWF is involved in platelet adhesion and prevents degradation of factor VIII; therefore, vWF deficiency or dysfunction impairs primary hemostasis as well as the intrinsic pathway of secondary hemostasis. In most cases, vWD is an inherited disorder caused by mutations in the vWF gene. Acquired vWD (aVWD) is rare and often occurs due to an underlying condition (e.g., malignancy, autoimmune disease, cardiovascular disorder). vWD may be asymptomatic or manifest with abnormal bleeding (e.g., epistaxis, heavy menstrual bleeding, prolonged bleeding after surgical procedures). Diagnosis is confirmed by low vWF antigen and/or activity levels. Pharmacological treatment is indicated for active bleeding, pre-procedure prophylaxis, and management of heavy menstrual bleeding. Treatment options for vWD include desmopressin and vWF/factor VIII concentrates. Management for avWD additionally includes investigations for the underlying cause and treatment based on the underlying cause.


© AMBOSS
Epidemiology
- Most common congenital bleeding disorder [1]
- Prevalence: estimated to affect approx. 1% of the US population [1]
Epidemiological data refers to the US, unless otherwise specified.
Etiology
| Variants of von Willebrand disease [2][3] | ||||
|---|---|---|---|---|
| Type | Description | Etiology | Mechanism | |
| Inherited von Willebrand disease 13576 | Type 1 (60–70%) [2] |
|
|
|
| Type 2 (20–30%) [2] |
|
|
||
| Type 3 (5–10%) [2] |
|
|
||
| Acquired von Willebrand disease (avWD) |
|
|
|
|
Pathophysiology
Deficiency or dysfunction of vWF leads to:
- Dysfunctional platelet adhesion → impaired primary hemostasis
- Reduced binding of factor VIII → increased degradation → ↓ factor VIII activity → impaired intrinsic pathway of secondary hemostasis

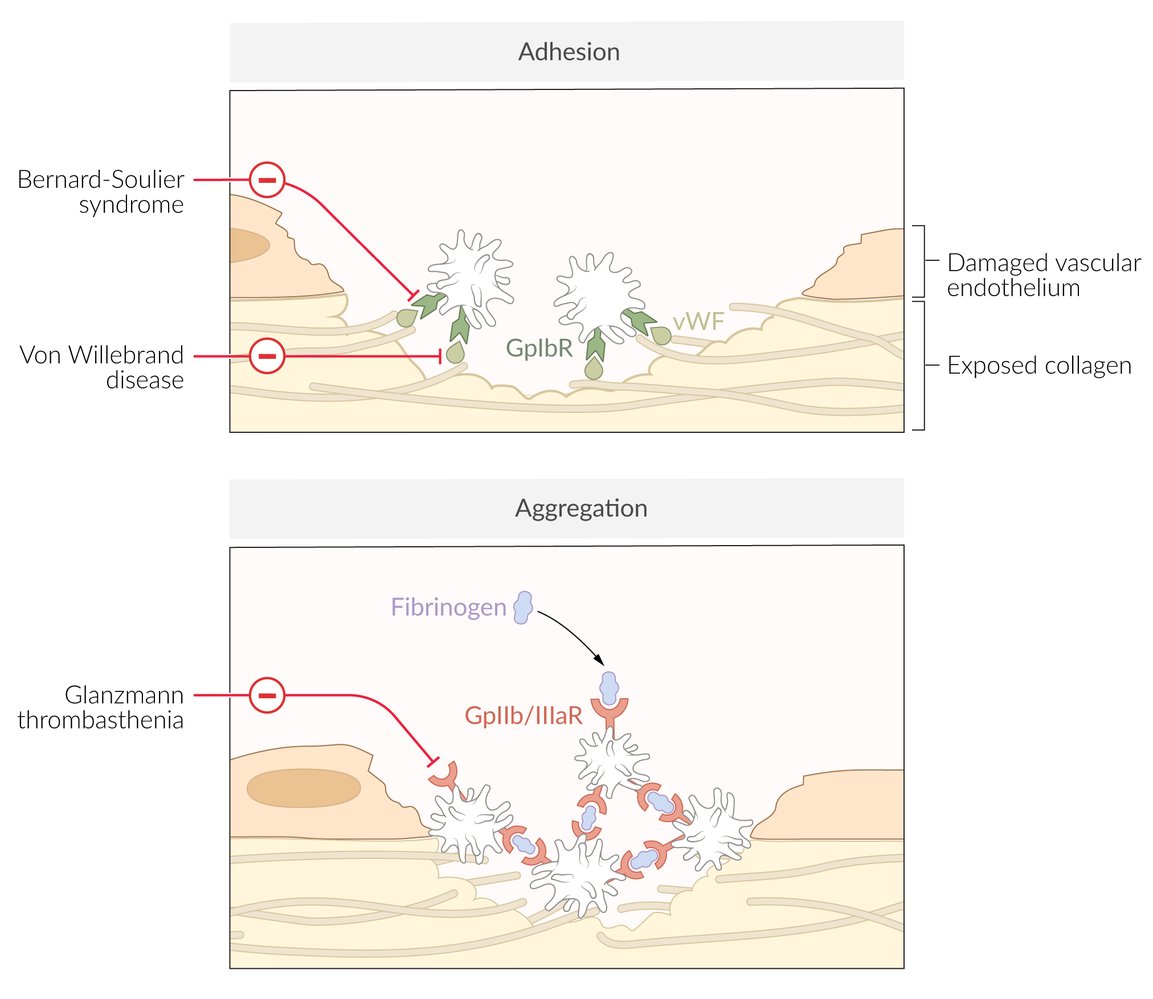
Red indicators link syndromes with their associated defective or absent molecules, according to which phase of primary hemostasis is affected (i.e., adhesion or aggregation)
© AMBOSS
Clinical features
Symptom severity varies between the different types of vWD. Type 1 and avWD usually have milder manifestations; type 3 is the most severe form. [2][4]
- May be asymptomatic
- Symptomatic individuals may develop the following symptoms:
-
Mucocutaneous bleeding
- Ecchymoses, easy bruising
- Epistaxis
- Bleeding of gingiva and gums
- Petechiae
- Prolonged bleeding from minor injuries
- Bleeding after surgical procedures or tooth extraction
- GI bleeding (can be caused by angiodysplasia)
- Heavy menstrual bleeding (affects up to 92% of women with vWD) [5]
- Postpartum hemorrhage
- Severe cases: large hematomas, hemarthrosis, life-threatening bleeding (e.g., during childbirth)
-
Mucocutaneous bleeding
Diagnosis
See also “Diagnostic workup of bleeding disorders” for a general workup of patients presenting with bleeding diathesis.
Approach [6]
- Suspect vWD in patients with:
- Abnormal bleeding
- A positive family history
- Bleeding assessment tools (BATs) can help assess the likelihood of vWD.
- Refer patients to hematology if there is high clinical suspicion for a bleeding disorder, even if BAT score is normal. [2]
- Routine laboratory studies (e.g., CBC, coagulation studies) may show supportive findings.
- vWD studies are required to confirm the diagnosis.
Inherited vWD typically manifests with recurrent bleeding episodes from childhood.
Initial studies
Routine laboratory studies [6][7]
-
CBC
- Platelet count: normal or ↓
- Hb: normal or ↓ (due to prolonged bleeding, e.g., heavy menses) [2]
-
Coagulation studies
- aPTT: normal or ↑ (due to factor VIII deficiency)
- PT: normal
- Mixing study (rarely performed in practice): In inherited vWD, a prolonged aPTT will normalize. [7]
- Ferritin: normal or ↓ (due to prolonged bleeding) [2]
Normal CBC and coagulation studies do not rule out vWD. [6]
vWD studies [2][6]
- Diagnosis requires the assessment of both:
- vWF antigen levels using a vWF antigen assay
-
Platelet-dependent vWF activity, e.g., using:
- vWF:GpIbR
- Ristocetin cofactor assay (measures platelet agglutination)
- vWD is diagnosed if vWF antigen levels and/or vWF activity are low (i.e., < 30 IU/dL; without bleeding or < 50 IU/dL with abnormal bleeding).
- Further studies can help confirm the diagnosis and identify the subtype.
- Factor VIII activity: may be low, e.g., in type 3 vWD, or normal
- Platelet-dependent vWF activity/vWF antigen ratio: may be low (< 0.7), e.g., in type 2 vWD, or normal
Factors such as pregnancy, hormonal therapy, and illness can affect vWD study results. If clinical suspicion for vWD is high, these studies should be repeated once the patient is back to their baseline. [2]
Advanced studies [2][6]
Subsequent studies are specialist-guided.
- Studies to confirm subtype of inherited vWD, e.g.:
- vWF multimer analysis
- vWF collagen binding/vWF antigen ratio
- Genetic testing
- Studies for avWD [3]
- Anti-vWF antibody: to help assess severity
- Evaluation for the underlying cause (e.g., malignancy, autoimmune diseases) based on the clinical picture
Management
General principles [2][8]
- Management is specialist-guided; consult hematology.
- The treatment approach is shaped by the clinical context, type of vWD, and vWD severity.
- Advise patients to avoid contact sports and/or activities with a high risk of head injury.
- Identify and treat the underlying cause of avWD. [3]
-
Indications for pharmacological treatment for vWD include: [9]
- Active bleeding
- Pre-procedure prophylaxis
- Manage complications (e.g., iron deficiency anemia).
- For patients with heavy menstrual bleeding: Consider hormonal treatment (e.g., oral contraceptives, progestin intrauterine device).
Platelet aggregation inhibitors (e.g., aspirin, NSAIDs, clopidogrel) should be used with caution in vWD because they further increase the risk of bleeding. [8]
Pharmacological treatment for vWD [2][8]
Inherited vWD
-
Desmopressin
-
Preferred initial agent in type 1 vWD and some individuals with type 2 vWD for:
- Mild to moderate active bleeding
- Prophylaxis for minor surgery or procedure
- Consider a desmopressin trial to confirm adequate response.
-
Preferred initial agent in type 1 vWD and some individuals with type 2 vWD for:
-
vWF/factor VIII concentrate
-
Used for: [2]
- Major bleeding or major surgery in any type of vWD
- Any active bleeding or pre-procedure prophylaxis when desmopressin is not effective or contraindicated
- May be considered as long-term prophylaxis for patients with severe, frequent bleeding
-
Used for: [2]
-
Antifibrinolytics (e.g., tranexamic acid) [8][9]
- Often used as an adjunct to desmopressin or vWF/factor VIII concentrate
- May be used alone as prophylaxis for very minor procedures or heavy menstrual bleeding [2][8]
Desmopressin stimulates the release of stored vWF from endothelial cells and is therefore not effective for type 3 vWD because there is no vWF to be released. [8]
Acquired vWD [3]
- Treatment of the underlying cause
- Desmopressin or vWF/factor VIII concentrate can be used, but they are often less effective than in inherited vWD.
- Consider immunomodulatory interventions (e.g., if the underlying disorder cannot be treated) [3]
- IVIg
- Plasmapheresis
Approach to active bleeding [2][8]
- For patients with severe bleeding: See "Hemorrhagic shock."
- Start pharmacological treatment for vWD.
- Identify and treat the source of bleeding (e.g., with upper endoscopy).
- For patients with heavy menstrual bleeding:
- Consider underlying gynecologic conditions.
- See also "Diagnosis of abnormal uterine bleeding."
External Resources
References
- "What is von Willebrand Disease?". https://web.archive.org/web/20200804235405/https://www.cdc.gov/ncbddd/vwd/facts.html. [2019-10-28]
- Seidizadeh O, Eikenboom JCJ, Denis CV, et al. "von Willebrand disease". Nat Rev Dis Primers. 10(1). (2024)
- Franchini M, Mannucci PM. "Acquired von Willebrand syndrome: focused for hematologists". Haematologica. 105(8). :2032-2037. (2020)
- "The Diagnosis, Evaluation and Management of von Willebrand Disease". https://www.nhlbi.nih.gov/sites/default/files/media/docs/vwd.pdf. [2007-12-01]
- "Committe Opinion: Von Willebrand Disease in Women". https://www.acog.org/clinical/clinical-guidance/committee-opinion/articles/2013/12/von-willebrand-disease-in-women#. [2013-12-01]
- James PD, Connell NT, Ameer B, et al. "ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease". Blood Adv. 5(1). :280-300. (2021)
- Neutze D, Roque J. "Clinical Evaluation of Bleeding and Bruising in Primary Care". Am Fam Physician. 93(4). :279-86. (2016)
- Connell NT, Flood VH, Brignardello-Petersen R, et al. "ASH ISTH NHF WFH 2021 guidelines on the management of von Willebrand disease". Blood Adv. 5(1). :301-325. (2021)
- Leebeek FWG, Eikenboom JCJ. "Von Willebrand’s Disease". N Engl J Med. 375(21). :2067-2080. (2016)