Summary
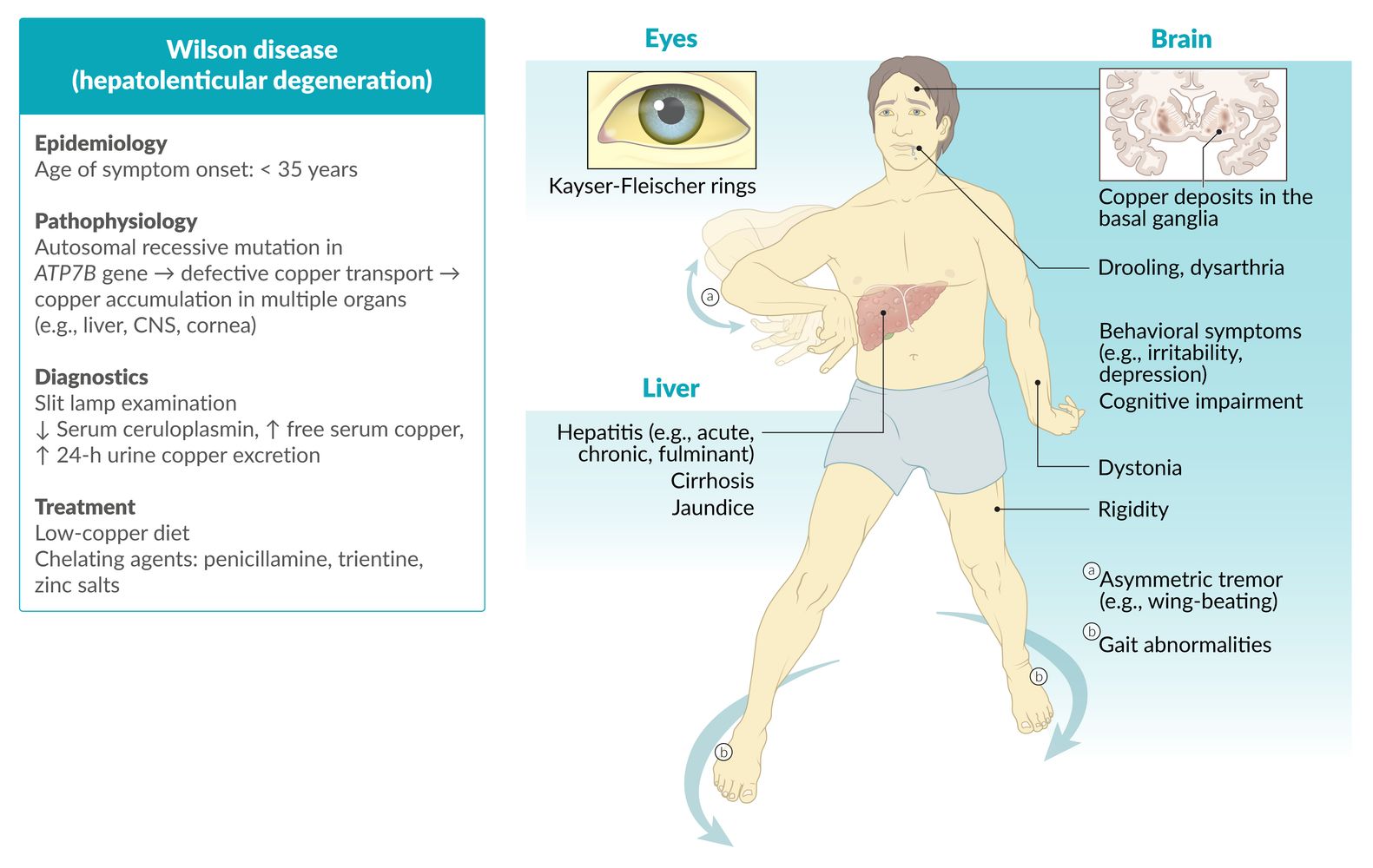
Wilson disease is an autosomal recessive metabolic disorder in which impaired copper excretion causes copper to accumulate in the body. Early-stage Wilson disease is characterized by the presence of copper deposits in the liver. As the disease progresses, copper accumulates in other organs as well, most importantly in the brain and cornea. The disease often goes undiagnosed until clinical suspicion is raised by the characteristic combination of hepatitis or cirrhosis, dementia, and parkinsonism. Kayser-Fleischer rings (brownish copper deposits visible around the iris) are a further indication of Wilson disease. Diagnostics involve slit lamp examination and laboratory studies to assess copper metabolism (e.g., ceruloplasmin, urinary copper excretion). Genetic testing or liver biopsies with quantitative copper assays may be necessary if the diagnosis is indeterminate. Management consists of maintaining a low-copper diet and administration of a chelating agent (e.g., penicillamine) or zinc salts. Individuals with Wilson disease have a good prognosis if the condition is diagnosed and treated early.


© AMBOSS
© AMBOSS
Epidemiology
- Age of onset: : 5–35 years ; (mean age 12–23 years) [1][2]
- Prevalence: ∼ 1/30,000 [3]
Epidemiological data refers to the US, unless otherwise specified.
Pathophysiology
-
Autosomal recessive mutations in the ATP7B gene (Wilson gene) on chromosome 13, which encodes for a membrane-bound, copper-transporting ATPase → defective ATP7B protein [4]
- Reduced incorporation of copper into apoceruloplasmin; → ↓ serum ceruloplasmin
- Reduced biliary copper excretion
- Results in ↑ free serum copper → accumulation in the liver, cornea, CNS (basal ganglia, brain stem, cerebellum), kidneys, and enterocytes [5]





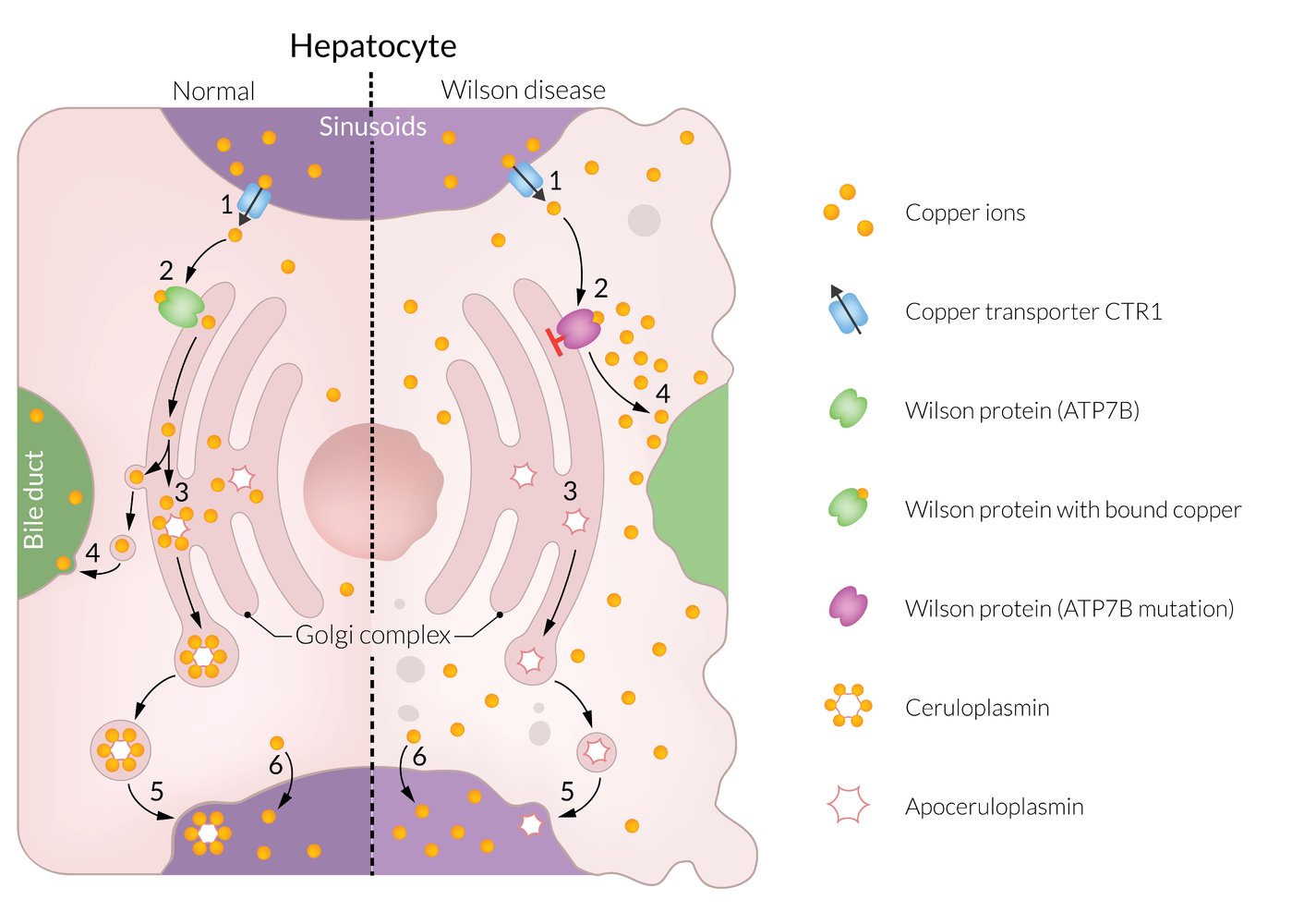
Left (physiologic):
(1) Copper ions are absorbed in the intestine and transported via the portal vein to the liver sinusoids, where they are taken up by the hepatocytes. (2) In the hepatocytes, copper ions enter the Golgi complex via the Wilson protein (ATP7B). (3) In the Golgi complex, the copper ions bind to apoceruloplasmin, thereby forming ceruloplasmin. (4) Excess copper is excreted into the bile via the Wilson protein. (5) Ceruloplasmin is secreted into plasma and spread throughout the body. (6) A small amount of unbound copper ions passes directly into the blood and is available as free serum copper.
Right (Wilson disease):
In Wilson disease, the Wilson protein is dysfunctional. Copper ions can neither be taken up by the Golgi complex (3) nor excreted into the bile (4). As a result, copper ions accumulate in the hepatocytes, which leads to hepatic damage. A large amount of excess, unbound copper ions passes directly into the bloodstream (6) raising the free serum copper concentration. This results in copper deposition in other organs.
© AMBOSS
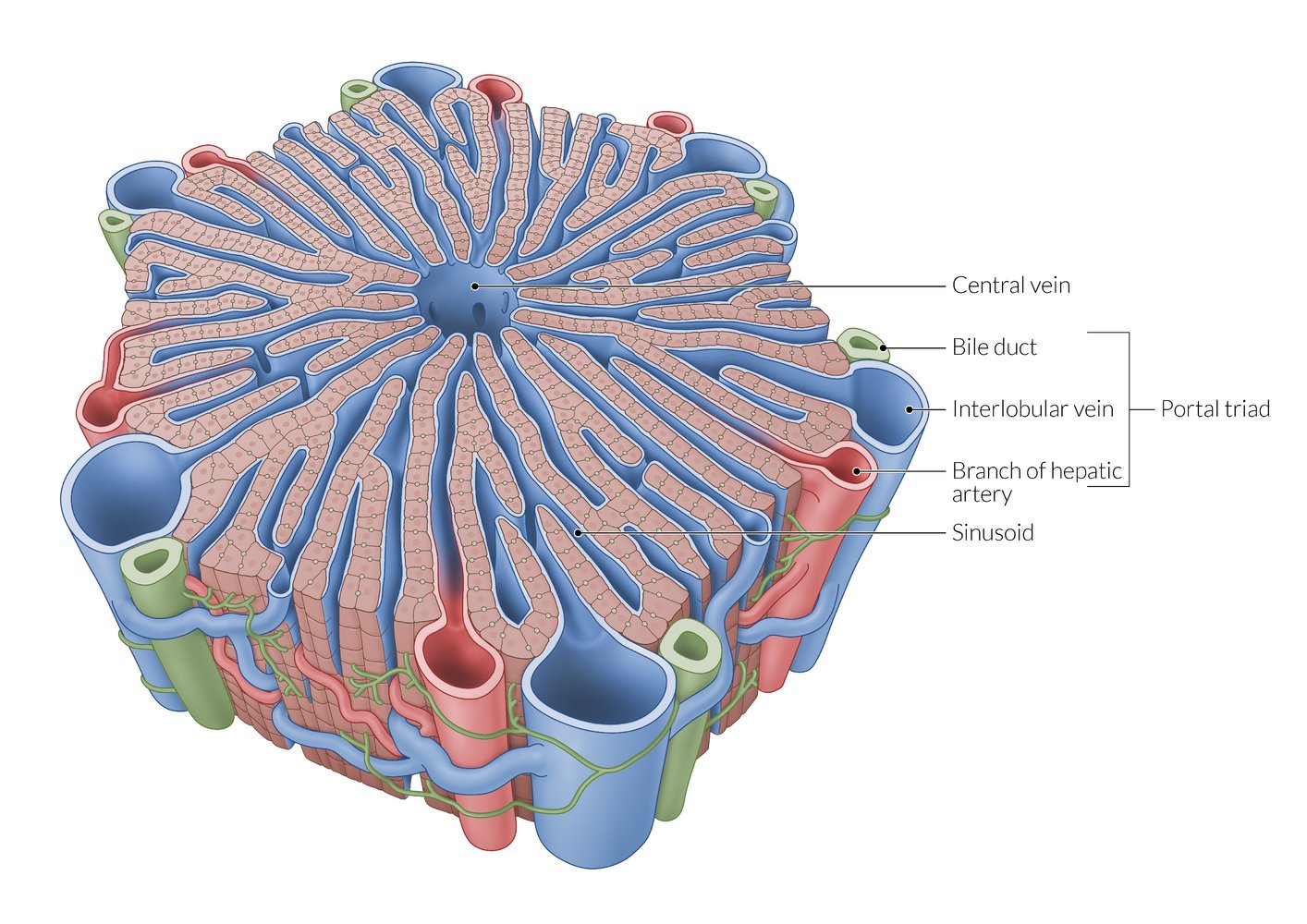
A lobule is a roughly hexagonal structural unit in the liver, separated from adjacent lobules by connective tissue. In each lobule, plates of hepatocytes are separated by hepatic sinusoids. At the vertices, branches of the hepatic artery and portal vein are arranged with bile ducts into portal triads. The flow of blood is from peripheral to central, as blood from the arteries and veins in the portal triads mixes in the sinusoids and drains via a central vein.
Note that sources differ regarding the distribution of portal triads, with some sources depicting them at every vertex of the hepatic lobules (as shown here) and others at only every second vertex.
© AMBOSS
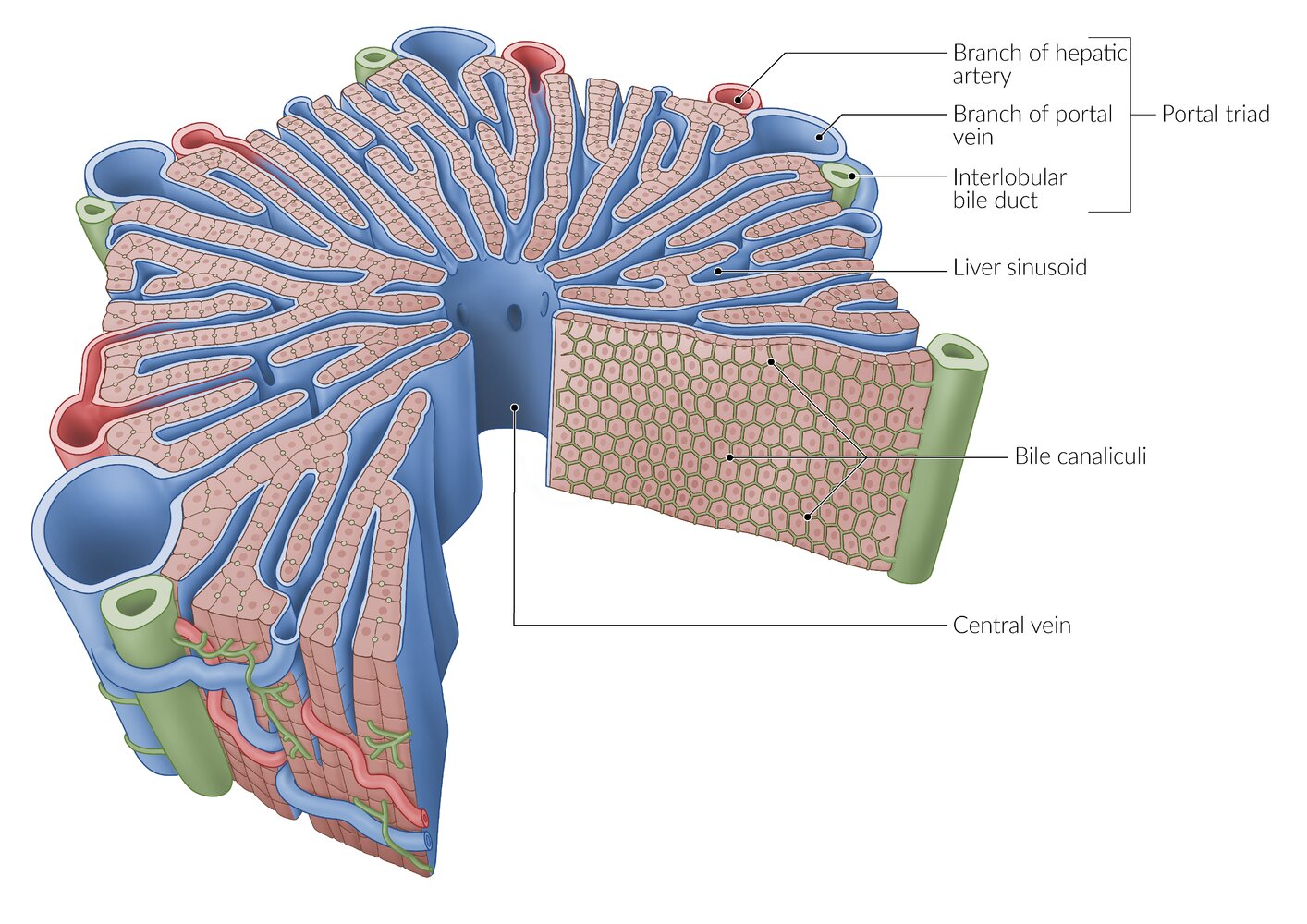
A lobule is a roughly hexagonal structural unit in the liver, separated from adjacent lobules by connective tissue. In each lobule, plates of hepatocytes are separated by hepatic sinusoids. At the vertices, branches of the hepatic artery and portal vein are arranged with bile ducts into portal triads. The flow of blood is from peripheral to central, as blood from the arteries and veins in the portal triads mixes in the sinusoids and drains via a central vein.
Between the hepatocytes is a network of bile canaliculi, which drains bile towards the interlobular bile ducts in the portal triads (the flow of bile is from central to peripheral, and thus opposite to the flow of blood).
© AMBOSS
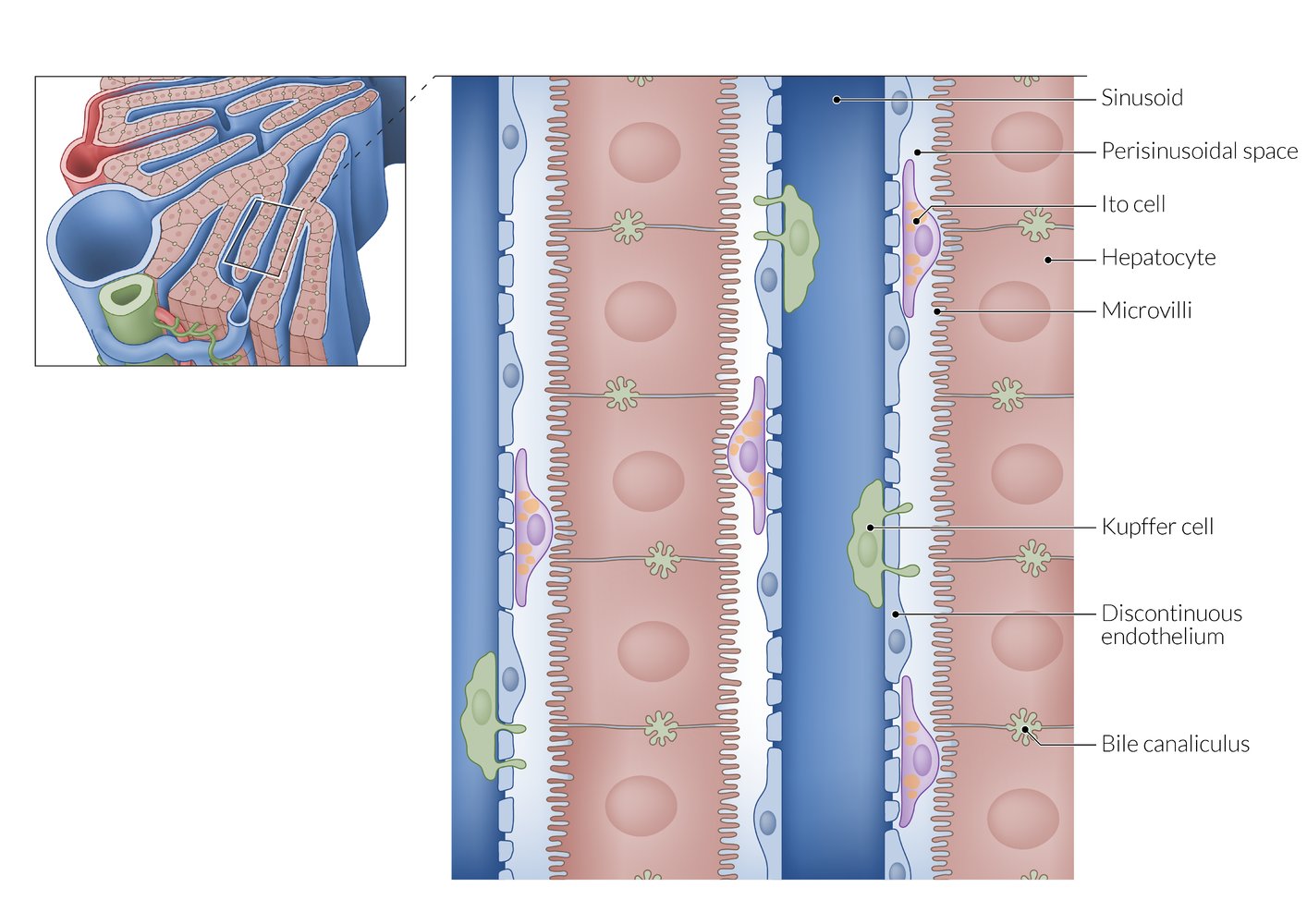
From the sinusoids, plasma can enter the perisinusoidal space through the highly fenestrated endothelium. Microvilli on the basolateral surface of the hepatocytes increase the surface area for efficient substance exchange. The apical surfaces of adjacent hepatocytes form the bile canaliculi, which drain into the peripherally located interlobular bile ducts.
© AMBOSS
© AMBOSS
Clinical features
-
Hepatic: variable degrees of liver disease possible
- Hepatosplenomegaly
- Jaundice
- Ascites
- Abdominal pain
- Symptoms of acute or chronic hepatitis (may be indistinguishable from autoimmune hepatitis)
-
Signs of chronic liver disease
- Hepatic encephalopathy
- Cirrhosis
- Portal hypertension
- Symptoms of acute liver failure
-
Ocular
-
Kayser-Fleischer rings
- Copper accumulation in the Descemet membrane; that results in 1–2 mm wide; golden or green-brown rings in the periphery of the cornea
- Characteristic for Wilson disease, but absence does not rule out the diagnosis
- Sunflower cataracts: copper deposits in the lens causing opacification in the shape of a sunflower
-
Kayser-Fleischer rings
-
Neurological
-
Cerebellar symptoms, e.g.:
- Dysarthria (most common)
- Gait abnormalities
- Choreoathetosis
-
Extrapyramidal symptoms, e.g.:
- Dystonia
- Parkinsonism
-
Tremor (usually asymmetric, affecting the hands), which may be any combination of:
- Resting tremor
- Intention tremor
- Wing-beating tremor: a low frequency, high amplitude tremor that is most prominent when the arms are outstretched anteriorly or laterally
- Drooling (caused by oropharyngeal dysphagia)
- Seizures
- Cognitive impairment
-
Cerebellar symptoms, e.g.:
-
Psychiatric and behavioral
- Major depressive disorder
- Irritability
- Psychosis
- Other: : e.g., Fanconi syndrome
Wilson disease should be suspected in cases of nonspecific noninfectious liver disease and in nonspecific extrapyramidal movement disorders.


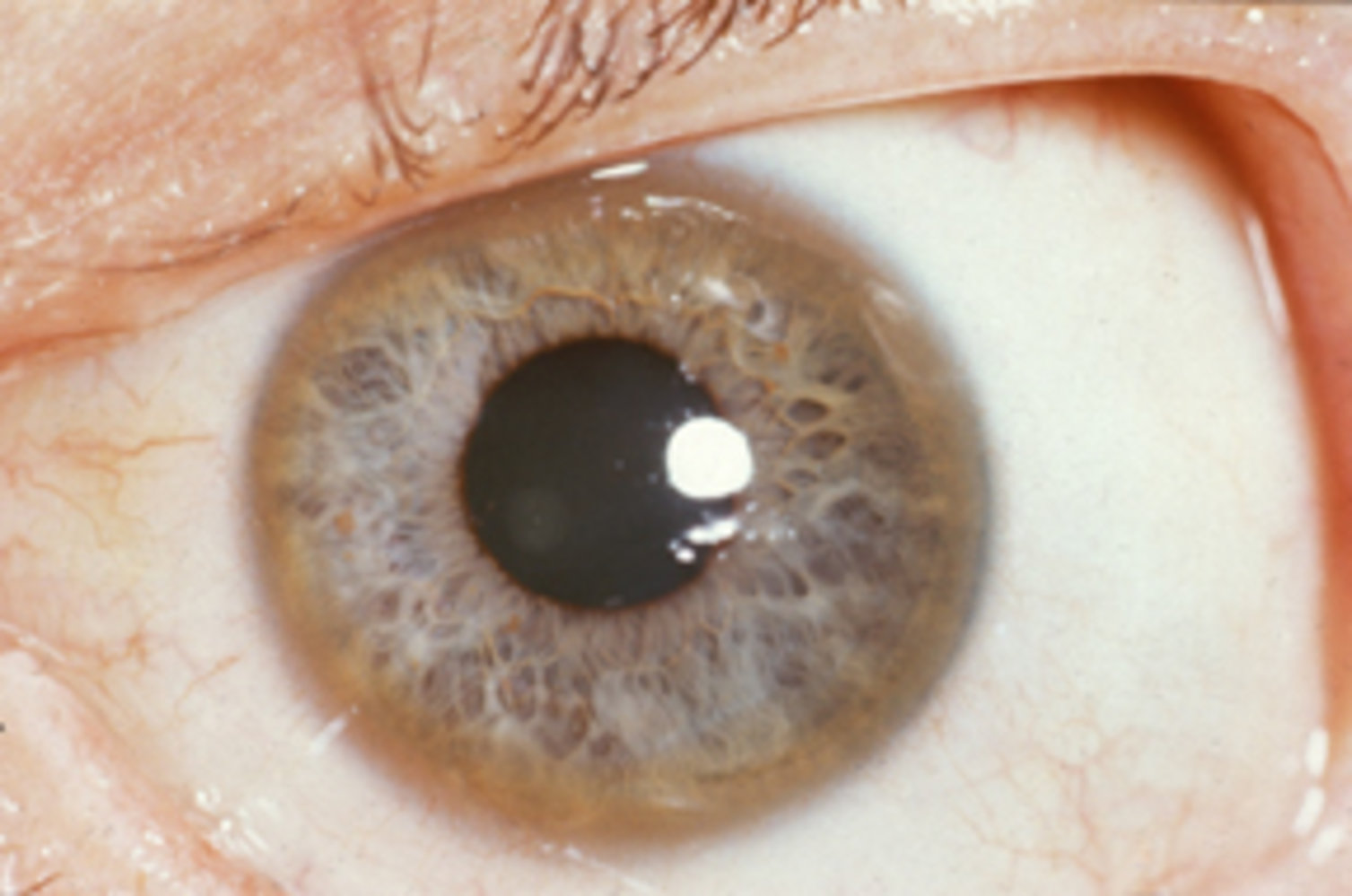
Slit-lamp photography of a left eye
A brown ring is visible in the periphery of the cornea (Kayser-Fleischer ring; green overlay).
Kayser-Fleischer rings are typically seen in Wilson disease.
Source: “Images of Memorable Cases: Case 9: Kayser-Fleischer_ring” by Herbert L. Fred, MD, Hendrik A. van Dijk, Wikimedia Foundation, licensed under CC BY 2.0. The supplementary image with overlays of relevant areas was adapted from the image mentioned above (© AMBOSS).
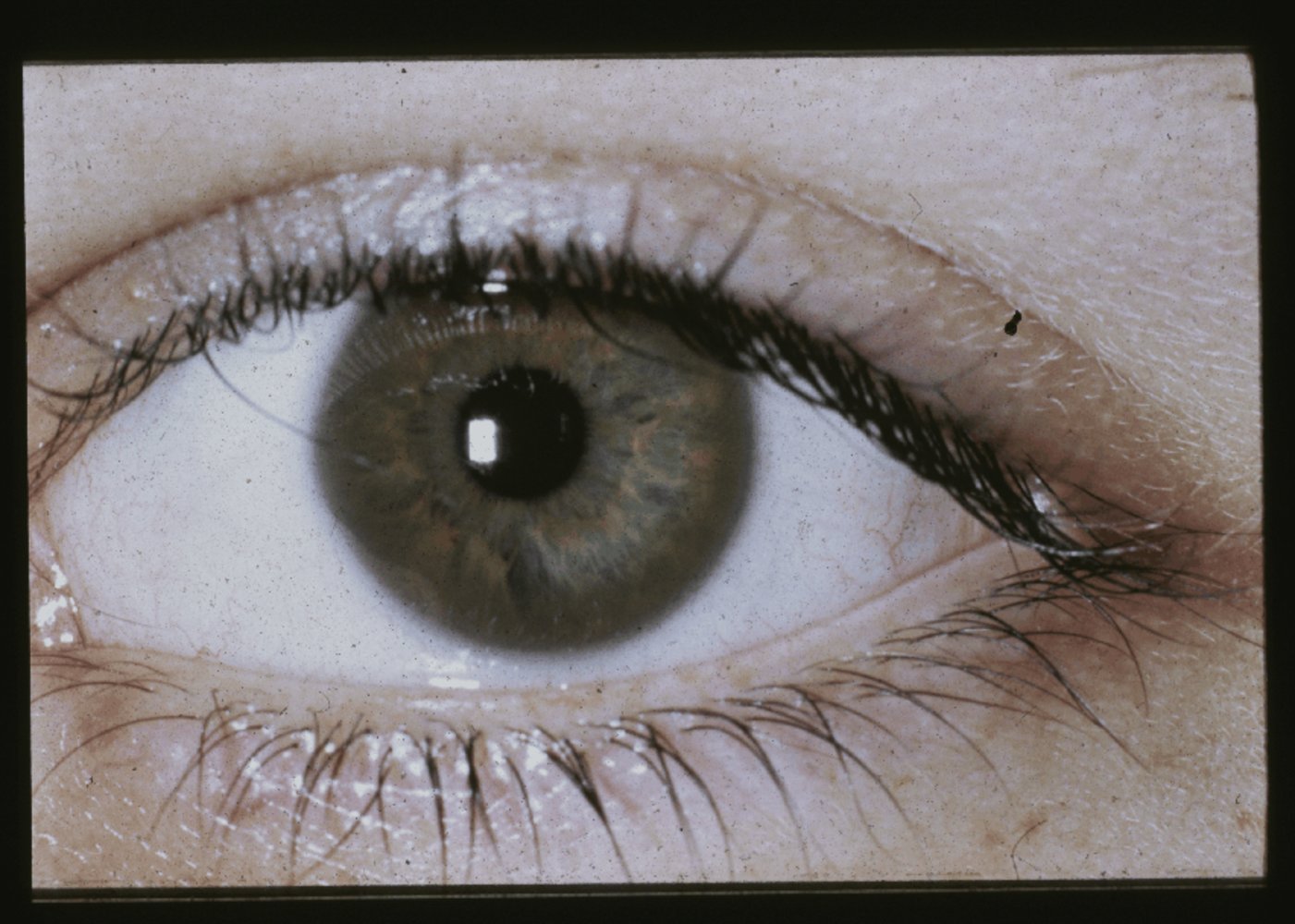
Slit-lamp photography of the left eye
A brown ring (Kayser-Fleischer ring; green overlay) is visible in the periphery of the cornea.
Kayser-Fleischer rings are characteristic of Wilson disease.
Source: "Case 1027 photo 1, clinical photo 1", David G. Cogan, Cogan Collection, NEI/NIH. licensed under Public Domain
Diagnosis
Approach [7]
- Consider a diagnosis of Wilson disease in individuals with:
- Acute liver failure (ALF) with nonimmune hemolytic anemia (i.e., negative Coombs test)
- Recurrent self-limited hemolysis
- Unexplained liver disease, especially if accompanied by neuropsychiatric symptoms
- A first-degree relative with Wilson disease
- Perform a comprehensive clinical assessment.
- Obtain routine laboratory studies to evaluate for liver involvement and hemolysis.
- Obtain copper metabolism studies to assess for Wilson disease.
- Consider advanced studies (e.g., liver biopsy, genetic testing) if the diagnosis remains unclear.
Routine screening for Wilson disease in patients with acute liver failure is not recommended. [8]
Clinical evaluation [6]
- Obtain a detailed personal and family history.
- Conduct a comprehensive examination, including:
- Neurological examination
- Psychiatric evaluation
- Slit lamp examination: Kayser-Fleischer rings, sunflower cataract
Laboratory studies
Routine laboratory studies [6]
-
Liver chemistries
- ↑ Transaminases; AST > ALT in cirrhosis
- ↑ Bilirubin, ↓ alkaline phosphatase in patients with ALF and/or hemolysis
- CBC: hemolytic anemia, thrombocytopenia, leukopenia may be present
- BMP: ↑ BUN, ↑ creatinine, ↓ potassium in renal involvement
-
Other
- Coombs test: negative
- ↑ INR in patients with severe liver damage
- ↓ Uric acid in patients with associated renal tubular dysfunction
- Urinalysis: may show microscopic hematuria and/or other findings consistent with Fanconi syndrome (e.g., aminoaciduria)
Copper metabolism studies [6]
- ↓ Serum ceruloplasmin (< 20 mg/dL)
- ↑ 24-hour urine copper excretion (> ULN)
-
Serum copper (optional)
- ↓ Total serum copper
- ↑ Free serum copper (i.e., non-ceruloplasmin-bound copper)
Interpretation of initial testing [6]
- In patients with one or more clinical features of Wilson disease:
- ↓ Serum ceruloplasmin and ↑ 24-hour urine copper: Diagnosis is confirmed.
- Inconclusive serum ceruloplasmin and/or 24-hour urine copper: Obtain advanced studies.
- Scoring systems (e.g., Leipzig score) may help establish the diagnosis.
The absence of typical findings (e.g., Kayser-Fleischer rings) does not exclude the diagnosis.
Advanced studies [2][7]
-
Liver biopsy: Consider if other tests are inconclusive. ; [2]
- May show positive copper staining (low sensitivity)
- Hepatic copper concentration > 250 mcg/g (dry weight) indicates Wilson disease. [7]
- Nonspecific findings include mild steatosis, fibrosis, and cirrhosis.
-
Genetic testing for ATP7B mutation in patients with either:
- First-degree relatives with Wilson disease
- Inconclusive diagnostic tests
-
MRI brain
- Consider in patients with neurological findings.
- Findings
- Hyperintensities in the basal ganglia, thalamus, pons, and white matter
- “Face of the giant panda” sign (rare but pathognomonic): a specific pattern of midbrain copper accumulation
Differential diagnoses
-
Hepatic
- Autoimmune hepatitis
- Viral hepatitis
- Hemochromatosis
- Nonalcoholic fatty liver disease
- Cirrhosis
-
Neuropsychiatric
- Parkinson disease
- Huntington disease
- Multiple sclerosis
- Schizophrenia
- Personality disorders
The differential diagnoses listed here are not exhaustive.
Treatment
General principles [6]
- Refer to hepatology for specialist-guided management.
- All patients require lifelong pharmacological therapy.
- Encourage a low-copper diet (e.g., avoidance of organ meats, shellfish, nuts, chocolate, copper-containing dietary supplements).
- Manage hepatic symptoms.
- Implement treatment of cirrhosis in advanced liver disease.
- Initiate management of acute liver failure in decompensated cirrhosis.
- Refer patients with ; refractory decompensated cirrhosis or acute liver failure for liver transplantation.
- Consult neurology and/or psychiatry for symptomatic management.
Pharmacological therapy [7]
-
Initial therapy
- First line: chelating agents; , e.g., penicillamine; (preferred) or trientine
- Alternative: zinc salts (may also be used first-line for asymptomatic individuals)
- Maintenance therapy: : reduced-dose zinc salts or a chelating agent
The goal of initial therapy is to eliminate copper; the goal of maintenance therapy is to prevent reaccumulation of copper.
Chelating agents should be titrated gradually. Rapid mobilization of the copper stored in tissues may exacerbate neurological symptoms.
Monitoring [6]
All patients should be regularly monitored for clinical and laboratory-based improvement and treatment side effects.
- Clinical assessment, including neurological examination, at least every 6 months
-
Laboratory studies
- At least every 6 months: serum copper, ceruloplasmin, liver enzymes, INR, CBC, and urinalysis
- At least once yearly: 24-hour urine copper
External Resources
References
- Lin LJ, Wang DX, Ding NN, et al. "Comprehensive analysis on clinical features of Wilson's disease: an experience over 28 years with 133 cases.". Neurol Res. 36(2). :157-63. (2014)
- European Association for Study of Liver. "EASL Clinical Practice Guidelines: Wilson's disease". J Hepatol. 56(3). :671-685. (2012)
- Amit Kulkarni, Vijay Kumar Sharma. "Wilson's Disease". Elsevier. :424-433. (2017). ISBN: 9780128037089
- Dong Q-Y, Wu Z-Y. "Advance in the pathogenesis and treatment of Wilson disease". Transl Neurodegener. 1. :23. (2012)
- Weiss KH, Zischka H. "Copper Directly Affects Intestinal Lipid Turnover". Gastroenterology. 154(1). :15-17. (2018)
- Schilsky ML, Roberts EA, Bronstein JM, et al. "A multidisciplinary approach to the diagnosis and management of Wilson disease: 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases". Hepatology. (2022)
- Schilsky ML, Roberts EA, Bronstein JM, et al. "A multidisciplinary approach to the diagnosis and management of Wilson disease: Executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases". Hepatology. (2022)
- Flamm SL, Yang Y-X, Singh S, et al. "American Gastroenterological Association Institute Guidelines for the Diagnosis and Management of Acute Liver Failure". Gastroenterology. 152(3). :644-647. (2017)
- Schilsky ML. "Wilson disease: Clinical manifestations, diagnosis, and natural history". UpToDate. UpToDate. https://www.uptodate.com/contents/wilson-disease-clinical-manifestations-diagnosis-and-natural-history?source=search_result&search=wilson%27s%20disease&selectedTitle=1~150#H57950462. [2015-11-10]
- Schilsky ML. "Wilson disease: Epidemiology and pathogenesis". UpToDate. UpToDate. https://www.uptodate.com/contents/wilson-disease-epidemiology-and-pathogenesis?source=see_link. [2015-01-13]
- Schilsky ML. "Wilson disease: Diagnostic tests". UpToDate. UpToDate. https://www.uptodate.com/contents/wilson-disease-diagnostic-tests?source=search_result&search=wilson%20disease&selectedTitle=2~150#H19. [2015-12-01]
- Schilsky ML. "Wilson disease: Treatment and prognosis". UpToDate. UpToDate. https://www.uptodate.com/contents/wilson-disease-treatment-and-prognosis#H26. [2016-11-29]